
基于煤气化细渣构建碳基氧还原催化剂及其催化性能研究
2022-08-01 05:46:44王思敏赵丽丽郭庆华于广锁
燃料化学学报 2022年6期
王思敏 ,龚 岩 ,李 恒 ,赵丽丽 ,郭庆华,* ,于广锁,2
(1. 华东理工大学 洁净煤技术研究所, 上海 200237;2. 宁夏大学 省部共建煤炭高效利用与绿色化工国家重点实验室, 宁夏 银川 750021)
随着煤气化技术的广泛应用,中国2020年煤气化渣产量已超3300万吨,其中,含碳量较高的气化细渣占比高达20%-40%[1]。目前,气化渣主要以堆存填埋处理为主,在造成大量碳资源浪费的同时也带来了严重的环境污染,气化渣的规模化和资源化利用已经成为煤气化企业亟需解决的关键问题。气化细渣因其滤饼含水量大、烧失量高和粒径分布跨度大等特点成为气化渣利用的难点问题[2]。浮选是气化细渣炭灰分离的有效手段之一,浮选得到的残炭结构稳定且含有丰富的碱金属和碱土金属[3],表面拥有大量的孔结构以及部分含氧官能团[4],浮选后残炭的这些优点使其满足作为氧还原碳基催化剂前驱体的基本要求[5,6]。对气化细渣利用的研究报道目前主要集中在返炉气化和锅炉掺烧,对气化细渣浮选残炭的高值化利用鲜有报道。因此,探究气化细渣构建氧还原催化剂的可行性,可促进气化细渣在新能源领域的高值化利用。在众多新能源研究方向中,燃料电池因转化效率高,供能稳定和环保效益好已使其成为解决目前能源困境的最有效手段[7,8]。
已有研究表明,燃料电池的阳极反应速率相对较高,而阴极上的氧还原反应(ORR)动力学缓慢,需要相当大的过电位克服反应过程中多步电子转移所需的能量,一般通过合适的催化剂降低其反应能垒,加快其反应速率[9]。目前应用最广泛的是Pt/C催化剂,但由于Pt/C催化剂成本高、稳定性差和易CO中毒失活等特性,使其在燃料电池中的应用受到较大制约[10]。因此,开发具有高催化活性,低成本且环境友好的氧还原催化剂对燃料电池的大规模商用具有重要的意义。
杂原子掺杂碳基催化剂具有成本低、稳定性高和原料来源广泛等优点,并且相较于Pt/C催化剂,其具有更好的耐甲醇交叉效应和抗CO性,是一种有效的Pt/C催化剂替代材料[5]。相较于其他杂原子,氮碳原子的尺寸相近,在氮原子取代碳原子的过程中,炭材料骨架结构的破坏程度较小,能够保持炭材料的稳定性[11,12],并且氮原子具有比碳原子更大的电负性,导致氮掺杂后邻近碳原子周围呈现相对较高的正电荷密度,改变了催化剂的表面电子构型,提供了更多的活性位点,这使得氮原子成为炭材料掺杂过程中最具前景的杂原子[13,14]。此外,氮掺杂炭材料会影响氧还原过程中O2分子的吸附模式,使O = O键更容易断裂,从而对碳基催化剂的ORR性能起到促进作用[10,15]。Jiang等[16]制备了以均匀共价有机框架纳米球作为前驱体的氮掺杂多孔碳基催化剂,通过对石墨化氮和吡啶氮的精确调控,使材料具有较好的氧还原催化性能,与商业Pt/C催化剂相当。Zhang等[17]采用两步炭化法制备了Co和N共掺杂碳基催化剂,该催化剂具有多级孔结构、高的局部有序度和高效Co-Nx活性结构,表征结果显示,该催化剂在碱性条件下,起始电位可达到1.00 V(vs RHE),半波电位为0.891 V,优于Pt/C催化剂。
本研究采用煤气化细渣浮选-酸洗后高炭为碳源,经过化学活化方法构建了碳基氧还原催化剂,结合SEM-EDS、XPS和拉曼光谱等表征,对比分析活化剂比例和氮源对所制碳基催化剂理化特性的影响规律。利用电化学工作站,对所制备催化剂的氧还原催化性能进行考察,进一步验证利用气化细渣制备氧还原催化剂的可行性。
1 实验部分
1.1 原料预处理
实验选用的碳源为经过浮选-酸洗后的某工业装置气化细渣中的高炭。由于浮选后的产物中仍有残留的灰分,使用盐酸(HCl,36%)进行酸洗脱灰处理。
对比气化细渣(FS)、浮选后残炭(AC)以及酸洗后高炭(HC),其工业分析和元素分析如表1所示,灰成分分析如表2所示。通过浮选-酸洗预处理,能够有效去除SiO2以外的无机物。

表1 原料的工业分析和元素分析Table 1 Proximate and ultimate analyses of raw materials
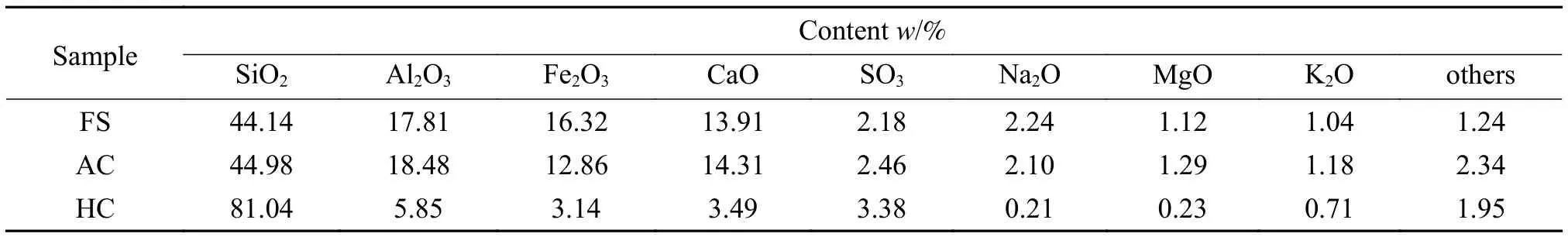
表2 原料的灰成分分析Table 2 Ash composition analysis of raw materials
1.2 材料制备
将预处理后的原料按照不同的比例与活化剂、氮源混合,将混合物放入石墨坩埚中置于卧式管式炉中在N2气氛下进行高温活化处理,升温速率为8 ℃/min,升温至 800 ℃,恒温 120 min,冷却至室温,用HCl洗涤后,用去离子水洗涤至中性,于烘箱中烘干备用。制备的不同条件下的催化剂材料分别为:
以KOH作为活化剂,NH4Cl作为氮源,以高炭∶KOH∶NH4Cl的配比分别为 1∶2∶3、1∶3∶3、1∶4∶3、1∶5∶3的比例进行催化剂材料制备,分别标记为CKN-123、CKN-133、CKN-143、CKN-153。
在使用KOH为活化剂的最佳条件下,替换氮源为三聚氰胺(C3H6N6)进行催化剂材料制备,标记为CKN6-143。
1.3 表征测试
通过扫描电子显微镜-能谱仪联用装置(SEMEDS,SU-510,日本 HITACHI)表征材料的微观结构以及进行表面元素分布的初步分析。采用X射线光电子能谱仪(XPS,ESCALAB 250Xi,美国 THERMO FISHER SCIENTIFIC)进一步表征材料表面元素组成、元素化学态及相对含量。利用激光显微拉曼光谱仪(inVia Reflex,英国RENISHAW)表征材料结构,拉曼激光器波长为532 nm,光谱分辨率为1 cm-1。使用物理吸附仪(ASAP-2020,美国MICR OMERITICS)在-196 ℃下进行氮气吸附实验,利用Brunauer-Emmett-Teller(BET)模型来计算材料比表面积和孔结构相关参数,分析孔径为2-200 nm。
1.4 电化学测试
利用旋转圆盘圆环电极装置(MSR,美国PINE)及电化学工作站(WaveDriver200,美国PINE)连用装置,利用循环伏安法(Cyclic Voltammetry,CV)、线性扫描伏安法(Linear Sweep Voltammetry,LSV)对材料的氧还原反应催化性能进行研究和分析。
实验采用三电极体系,使用的参比电极为银/氯化银电极(Ag/AgCl),对电极为铂丝(Pt),工作电极为旋转圆盘电极(RDE)或旋转环盘电极(RRDE),测试时,将制备的催化剂材料均匀地涂覆在工作电极表面。测试过程中使用的膜溶液为Nafion/乙醇溶液(10%),电解质为0.1 mol/L的氢氧化钾(KOH)溶液。
实验电位为1-0 V(vs RHE),CV测试的电势扫描速率为100 mV/s,LSV测试的电势扫描速率为10 mV/s,RDE选取的转速为225-1600 r/min,RRDE选取的转速为1600 r/min。
电子转移数(n)和对应的过氧化氢产率(H2O2,%)可以通过下式(1)和(2)计算。
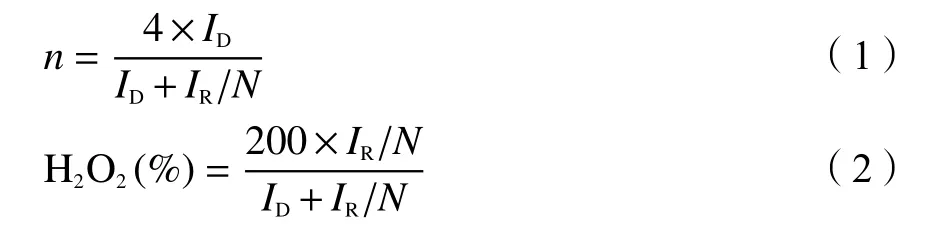
式中,IR和ID分别表示环电流和盘电流。N值表示电流收集效率,取0.377。
通过Koutecky-Levich(K-L)线性拟合得到动力学电流密度。
2 结果与讨论
2.1 活化剂比例对氧还原性能的影响
2.1.1 理化特性表征
通过SEM-EDS对材料的表面形貌和元素分布进行分析,如图1所示。对比图1(a)-(e)可以看出,材料经活化后孔隙结构更为明显,随着KOH添加量的增加,孔隙逐渐扩大,数量也逐渐增多。在高炭∶KOH∶NH4Cl=1∶4∶3时,材料 CKN-143表现出最优的孔隙形态,多为网络状孔,孔隙相对较大,孔壁较厚。如图1(e)所示,继续添加KOH,孔壁开始变薄,部分孔隙由于孔壁的坍塌连接在一起。KOH在活化过程中与炭发生反应,随着炭的消耗,孔隙发生显著变化,随着KOH添加量的增加,KOH的活化作用也会增强,孔的数量和相应的比表面积都会增加,当达到最优的孔隙度之后,若继续添加KOH则会产生过度侵蚀,导致孔隙结构的破坏。图1(f)为CKN-143的EDS分析结果,材料上存在着明显的N元素分布,可以初步判断活化过程中N掺杂效应明显。

图1 (a)气化细渣,(b)CKN-123,(c)CKN-133,(d)CKN-143,(e)CKN-153 的扫描电镜照片和(f) CKN-143 的 EDS 能谱照片Figure 1 SEM images of (a) gasification fine slag, (b) CKN-123, (c) CKN-133, (d) CKN-143,(e) CKN-153 and EDS spectrum of (f) CKN-143
材料的拉曼光谱谱图如图2所示,在1352和1588 cm-1处存在明显的特征峰,分别为D峰和G峰,两者的比值(ID/IG)可用来评估碳基催化剂的石墨化程度[5,18]。高石墨化程度的炭材料具有更好的稳定性和导电性[19]。由图2可知,CKN-143的ID/IG最低,约为1.01,表明其石墨化程度最高。CKN-123、CKN-133和CKN-153的ID/IG分别为1.05、1.03和1.05。气化细渣浮选残炭性质稳定,石墨化程度较高。在低KOH用量时,KOH对炭微晶有序排列的破坏效应仅体现在炭层边缘。同时,在高温下,KOH生成金属钾嵌入炭基体,碳原子发生重新排列和自组装导致材料形成类石墨化结构[20],材料总体石墨化程度略微上升。随着KOH用量的增加,KOH对炭微晶的刻蚀逐渐扩大到基体空间层面,导致炭微晶有序排列的破坏程度加深,使得材料总体的石墨化程度随着KOH用量的增加而降低。
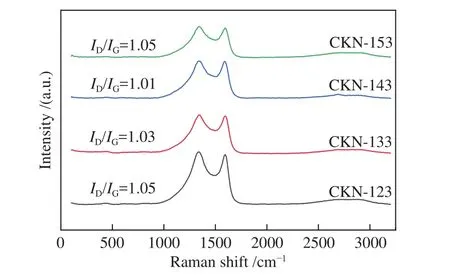
图2 拉曼光谱谱图以及ID/IGFigure 2 Raman spectra and ID/IG
2.1.2 氧还原反应催化性能
图3(a)为 CKN-123、CKN-133、CKN-143和 CKN-153在通入饱和O2的0.1 mol/L KOH电解液中的循环伏安扫描(CV)曲线。材料均存在明显的氧还原峰,具备一定的氧还原性能。其中,CKN-143和CKN-153的峰值电位最高,为0.61 V(vs. RHE),CKN-123和CKN-133的峰值电位分别为0.58 V(vs.RHE)、0.59 V(vs. RHE)。进一步研究材料的ORR性能,利用RRDE对材料进行了DECV测试,如图3(b)所示。CKN-143的起始电位和半波电位分别为0.81 V(vs. RHE)和0.70 V(vs. RHE),极限扩散电流密度为4.90 mA/cm2,优于另外三种,即CKN-143具有更小的过电位,同时能提供更大的放电电流密度。通过式(1)、式(2)计算出催化剂材料的电子转移数和对应的过氧化氢产率,如图3(c)所示。通过计算,CKN-143在0-0.7 V(vs. RHE)的平均电子转移数为 3.71,大于 CKN-123(3.16)、CKN-133(3.50)和 CKN-153(3.66),说明 CKN-143 能够催化更多的4电子反应,产生的H2O2也更少。

图3 不同活化比例的材料在O2饱和的0.1 mol/L KOH溶液中(a)CV曲线,(b)旋转环盘电极DECV曲线和(c)电子转移数与对应H2O2产率Figure 3 (a) CV curves, (b) DECV curves of RRDE and (c) electron transfer number and corresponding H2O2 yield of catalysts with different activation ratios in O2-saturated 0.1 mol/L KOH solutions
如图4(a)-(d)所示为四种材料在不同转速下的LSV曲线。不同材料均表现出明显的特征动力学区域。当电位高于0.8 V(vs. RHE)时,不同转速下电流密度基本一致,此时反应属于电化学极化区域,氧还原反应完全受电极反应速度控制。随着电位的逐渐降低,电流密度迅速升高,反应进入电化学极化与浓差极化的混合控制区。当电位下降至0.6 V(vs. RHE)时,曲线下降变缓,此时反应逐渐进入浓差极化区域。随着转速上升,传质增强,此区域内的电流密度也相应上升,进一步证明了此时电解液中氧气传输到催化剂层表面的速率成为反应的主要限制因素。对比图4(a)-(d)还发现,CKN-143的LSV曲线在浓差极化区域具有明显的平台,极限电流的平台越平,说明反应越趋于4电子反应,此结果与平均电子转移数的计算结果也吻合。
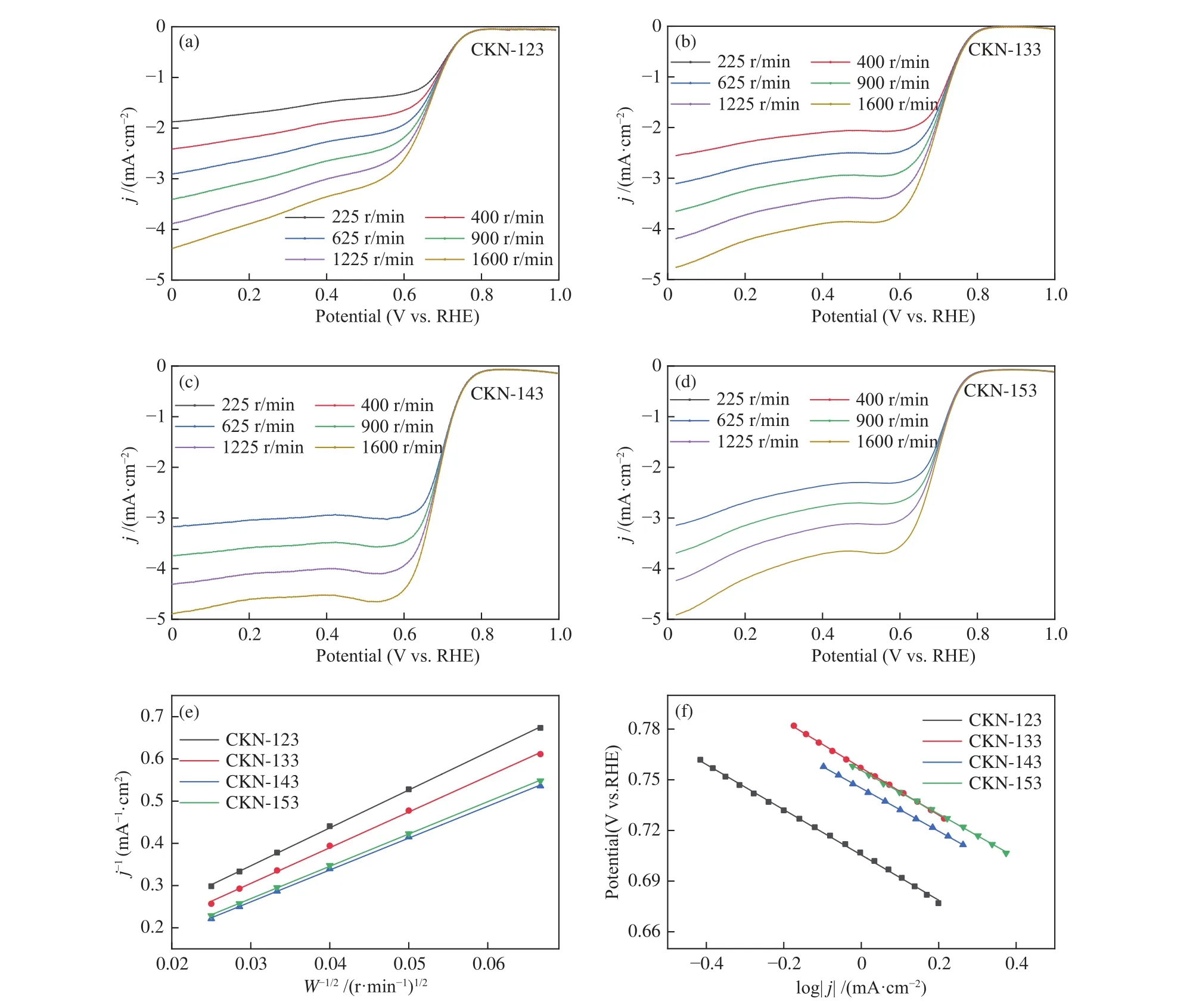
图4 O2饱和的 0.1 mol/L KOH 溶液中,(a)CKN-123、(b)CKN-133、(c)CKN-143 和(d)CKN-153 在不同转速时的 LSV 曲线、(e)K-L 曲线和(f)Tafel曲线Figure 4 In O2-saturated 0.1 mol/L KOH solutions, LSV curves of (a) CKN-123, (b) CKN-133, (c) CKN-143 and(d) CKN-153 at different rotation rates, (e) K-L curves and (f) Tafel curves
通过线性拟合获得材料的K-L曲线,如图4(e)所示。根据拟合结果得到对应的动力学电流密度,其中,CKN-143的动力学电流密度jk为28.08 mA/cm2,大于 CKN-123(12.92 mA/cm2)、CKN-133(19.54 mA/cm2)和 CKN-153(25.47 mA/cm2),CKN-143具有更优的动力学属性。
图4(f)为拟合的四种材料的Tafel曲线,对应Tafel斜率分别为:133.87 mV/dec(CKN-123)、139.43 mV/dec(CKN-133) 、 127.59 mV/dec(CKN-143)和128.90 mV/dec(CKN-153)。CKN-143 的 Tafel斜率小于另外三种,说明CKN-143在催化氧还原反应过程中具有更小的电子转移阻力,电化学过程中的能量损失也更低[21,22]。
总之,随着活化剂KOH添加量的增加,材料的氧还原催化性能呈现先增强后减弱的变化趋势,高温条件下,KOH适量添加能有效地调节材料的孔隙结构,促进石墨化有序发展,增强导电性,配合渗氮,氧还原催化性能得到增强[20,23]。当高炭和KOH质量比达到1∶5时,过量的KOH反而会破坏孔结构和石墨化程度,进而导致材料电导率的降低,影响催化性能。
2.2 不同氮源对氧还原性能的影响
选取活性最优的CKN-143,更换氮源为三聚氰胺制备催化剂材料,记为CKN6-143。探究不同渗氮剂对材料性能的影响,并对CKN-143和CKN6-143两种材料进行相应的表征。
2.2.1 理化特性表征
从扫描电镜照片(图5)可以看出,相较于CKN-143,CKN6-143在保持孔壁完整的同时具有更加丰富的孔隙结构,并没有出现过度烧蚀后孔壁断裂的情况,说明氮源不同也会对材料的孔隙结构有显著影响。

图5 (a)CKN-143 和(b)CKN6-143 的扫描电镜照片Figure 5 SEM images of (a) CKN-143 and (b) CKN6-143
图6为两种材料的拉曼光谱谱图,活化后,两种材料的石墨化程度都有了明显的提高。CKN6-143的ID/IG为0.98,低于CKN-143,表明CKN6-143的石墨化程度更高。

图6 CKN-143和CKN6-143的拉曼光谱谱图Figure 6 Raman spectra of CKN-143 and CKN6-143
为了进一步探究氮掺杂后材料的化学性质,采用XPS对材料的化学组成及化学态进行表征,图7(a)为两种材料的XPS总谱图,两种材料均存在明显的的N 1s谱峰。改变氮源后,氮含量从3.31%提高到了9.09%,说明三聚氰胺的氮掺杂效应更强。图7(b)-(e)为 CKN-143 和CKN6-143 的 C 1s和N 1s的高分辨率谱图。对C 1s峰进行分峰拟合后可得到三个拟合峰,分别对应C-C、C = O和C-N,C-N键的存在证明活化过程中完成了氮掺杂。进一步对氮的化学态进行分析,如图7(c)、(e)所示,N 1s谱图分为四个峰,在两种材料中,氮的存在形式均以吡啶氮(Pyridinic N)、吡咯氮(Pyrrolic N)、石墨氮(Graphitic N)和氧化氮(Oxidized N)形式存在,但其各自含量不同。通常认为,吡咯氮、吡啶氮和石墨氮这三种含氮官能团是氧还原反应的有效活性位点,可以加快氧还原反应速率,提高多孔炭材料的电催化效果[5]。相比之下,CKN6-143中这三种有效官能团占总含氮量的比例较高(83.74%),其产生的催化效果也更强。
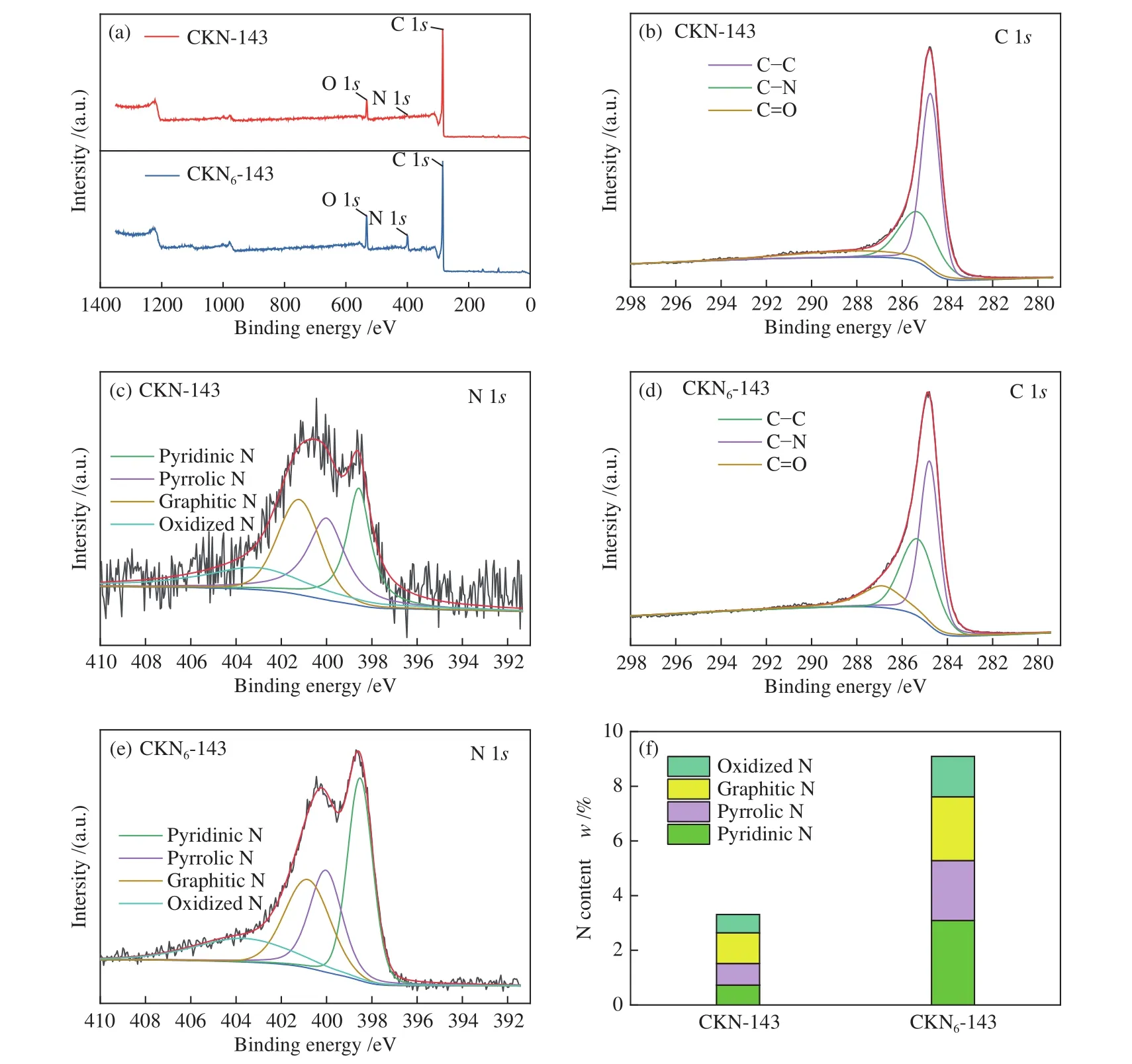
图7 (a)XPS 全谱图,(b)CKN-143、(d)CKN6-143 的 C 1s高分辨率谱图和(c)CKN-143、(e)CKN6-143 的 N 1s高分辨率谱图、(f)氮含量Figure 7 (a) XPS full spectra, C 1s high resolution spectra of (b) CKN-143, (d) CKN6-143 and N 1s high resolution spectra of (c)CKN-143, (e) CKN6-143, (f) content of N-containing species
图8(a)为两种材料的吸附-脱附曲线,均为IUPAC分类的Ⅳ型曲线,中间段出现H4型吸附回滞环,是Ⅰ型和Ⅱ型吸附等温线的复合,多出现在微孔和中孔混合的材料中,同时含有一些狭窄的裂隙孔。通过BET模型计算得出CKN6-143的比表面积略小于CKN-143(表3)。由孔径分布(图8(b))可知,两种材料都是微孔-介孔混合,大孔贡献极少。通过BJH孔容计算,尽管CKN-143的总孔容大于CKN-143,但CKN6-143的介孔孔容反而更大,介孔有利于离子的快速扩散,增强介质与内部孔隙之间的可达性,提供大量的有效表面,有利于材料催化性能的提高[24]。
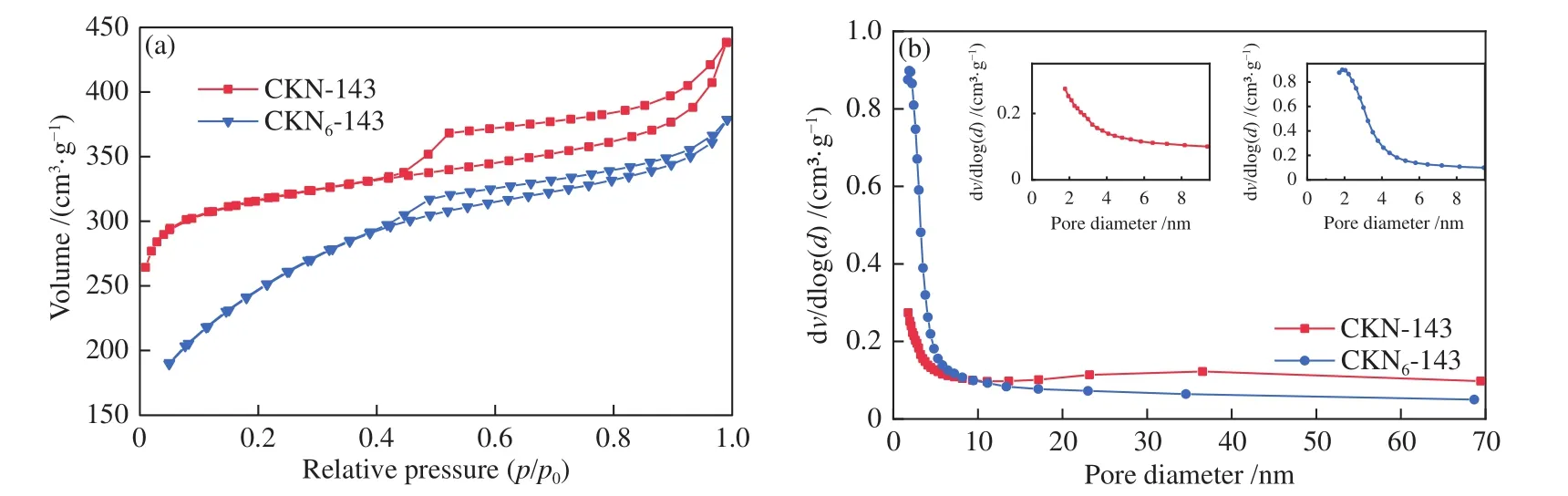
图8 CKN-143 和 CKN6-143 的(a)吸附-脱附曲线,(b)孔径分布Figure 8 (a) Nitrogen isothermal adsorption and desorption curves, (b) the pore size distribution of CKN-143 and CKN6-143
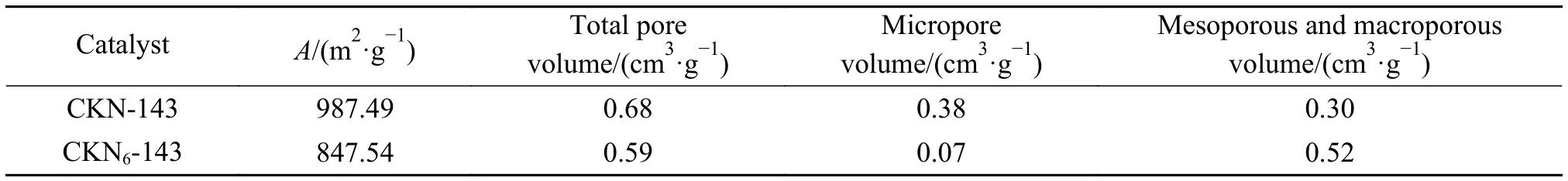
表3 CKN-143和CKN6-143的比表面积和孔容Table 3 Specific surface area (SSA) and pore volume of CKN-143 and CKN6-143
2.2.2 氧还原反应催化性能
利用电化学测试系统,对比两种材料的氧还原催化活性。图9为两种材料各项电化学性质曲线对比。由图9(a)、(b)可知,CKN6-143的CV 峰值电位为0.71 V(vs. RHE),较 CKN-143(0.61 V vs.RHE)有明显的提升。其起始电位和半波电位也比CKN-143高,分别为0.87 V(vs. RHE)和0.76 V(vs. RHE),其显著提升表示相比于CKN-143,CKN6-143的氧还原催化性能明显增强。两者的极限扩散电流密度没有明显差异,但由于CKN6-143的起始电位更高,即单位时间内,相同面积通过同样电流所需的过电位更小,所以催化活性也更高。由图9(c)-(e)可知,CKN6-143 在 0-0.7 V(vs. RHE)的平均电子转移数为3.82,平均过氧化氢产率为9.05%,与CKN-143相比,CKN6-143可以催化更多的4电子反应。通过线性拟合计算出的CKN6-143动力学电流密度jk为28.85 mA/cm2,优于CKN-143。由图可知,CKN6-143的起始电位和电子转移数略低于商用Pt/C,但CV峰值电位以及动力学电流密度则优于Pt/C,CKN6-143比CKN-143更加接近商用Pt/C的性能。
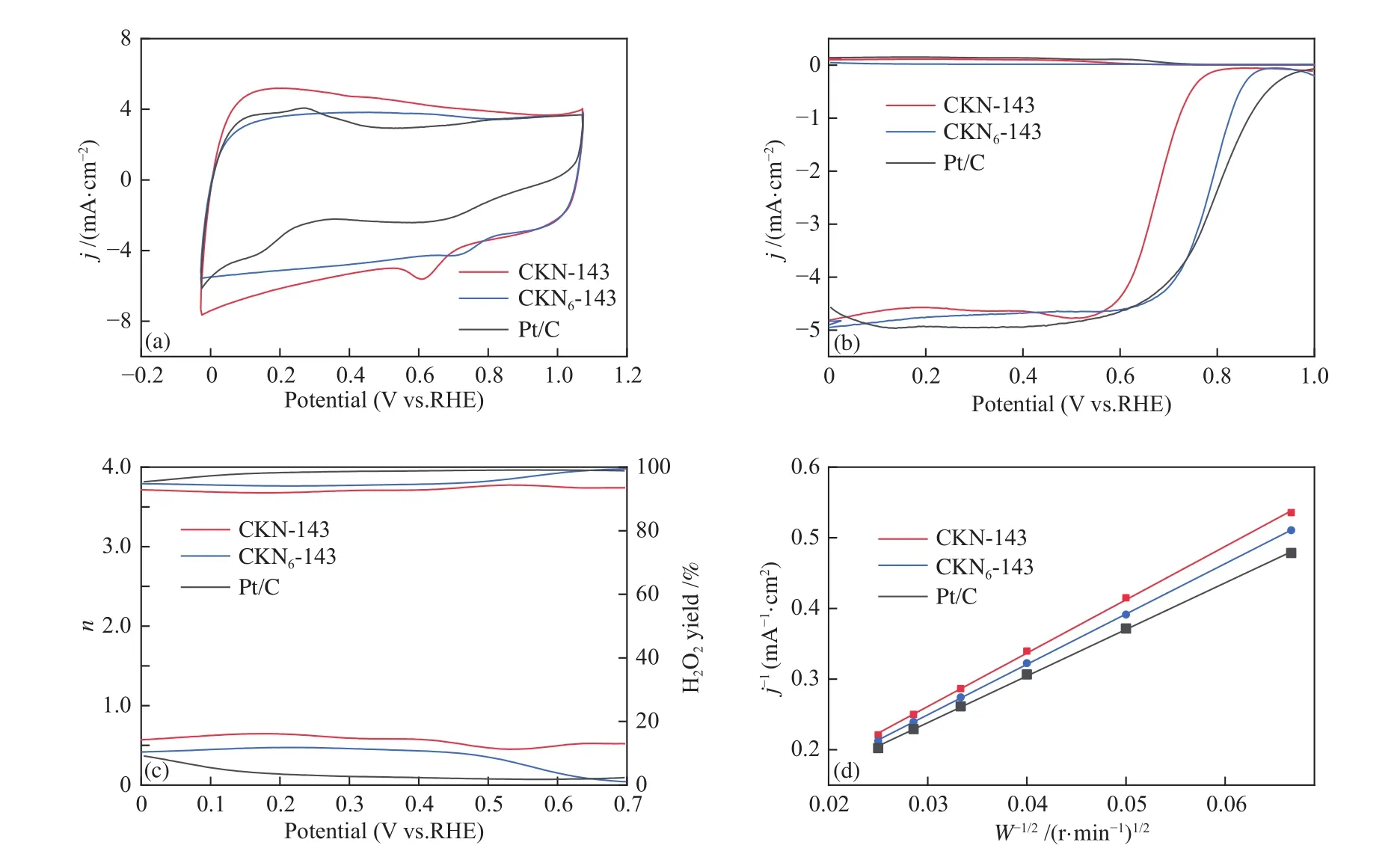
图9 在O2饱和的0.1 mol/L KOH溶液中,CKN-143,CKN6-143和Pt/C的(a)CV曲线,(b)旋转环盘电极DECV曲线,(c)电子转移数与对应 H2O2产率,(d)K-L 曲线Figure 9 In O2-saturated 0.1 mol/L KOH solutions, (a) CV curves, (b) DECV curves of RRDE, (c) electron transfer number and corresponding H2O2 yield, (d) K-L curves of CKN-143, CKN6-143 and Pt/C
为进一步研究催化剂的稳定性,将CKN-143,CKN6-143负载在碳纸上作为阴极催化材料,组装了用于测试的锌-空气电池。利用单电池进行循环充放电性能测试。由图10可知,两种材料组成电池的最终充电电压均为2.095 V左右,对比于初次循环的起始充电电压(2.175 V)仅下降了约3.7%,电池表现出良好的稳定性。而放电电压稳定在0.95 V,分别于1600和3000 min出现明显下降,CKN6-143的稳定性要优于CKN-143。
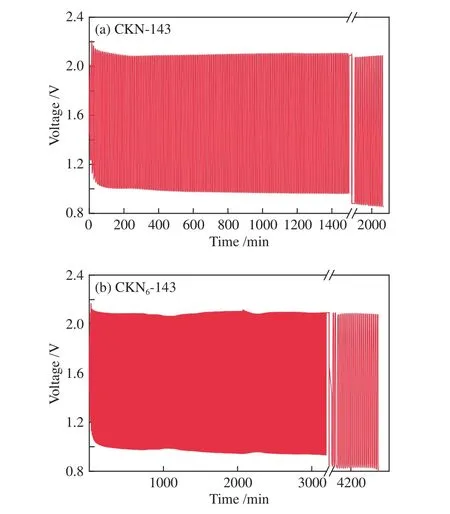
图10 基于CKN-143和CKN6-143在恒定充放电电流密度为10 mA/cm2时的锌空气电池的循环充放电性能Figure 10 Charge-discharge cycling performance of the Zn-air flow batteries based on CKN-143 and CKN6-143 catalysts at a constant charge-discharge current density of 10 mA/cm2
CKN6-143在所制备的氮掺杂碳基催化剂中表现出最为优异的氧还原催化性能,这一方面归因于CKN6-143具有良好的孔隙结构和高石墨化程度。材料催化性能不由材料的比表面积完全控制,介孔对于性能的影响更为关键。发达的介孔增强了离子的扩散,提供了大量的有效表面,石墨化程度高使得材料的导电性良好;另一方面,CKN6-143中较高程度的氮掺杂会改变碳原子周围的电荷密度和自旋密度,有助于产生更多的活性位点[25,26]。即三聚氰胺作为氮源,比NH4Cl更能促进碳基催化剂的氧还原性能。
3 结 论
本研究以煤气化细渣浮选-酸洗后高炭为碳源,经过化学活化方法制备了碳基氧还原催化剂,结合SEM-EDS、XPS和拉曼光谱等表征手段和电化学工作站,对比分析了活化剂比例和氮源对所制碳基催化剂理化特性的影响规律,建立了气化细渣构建碳基氧还原催化剂的基本方法。研究结果表明:
KOH作为活化剂对酸洗后高炭有明显的活化作用,适量的KOH能调节材料的孔结构,促进石墨化发展,提高电解质离子的传输效率。当高炭和KOH的质量比达到1∶5时,过量的KOH会导致孔结构被破坏,引起材料催化性能的下降。
不同氮源的氮掺杂效应不同,三聚氰胺作为氮源,比NH4Cl具有更强的氮掺杂效应,材料表面的氮含量从3.31%提高到了9.09%。
酸洗后高炭经过活化渗氮处理,具有良好的氧还原电催化性能,在高炭∶KOH∶三聚氰胺质量比为1∶4∶3的条件下制备出的材料具有较大的比表面积、较好的孔隙结构以及较高的氮含量。其起始电位可达0.87 V(vs. RHE),极限扩散电流密度为4.95 mA/cm2,平均电子转移数为3.82,具有较好的电化学性能。
猜你喜欢
环境卫生工程(2021年4期)2021-10-13 06:51:58
电镀与环保(2017年5期)2017-12-19 12:06:09
电镀与环保(2016年3期)2017-01-20 08:15:32
电镀与环保(2016年3期)2017-01-20 08:15:28
电镀与环保(2016年2期)2017-01-20 08:15:23
中国酿造(2016年12期)2016-03-01 03:08:11
氮肥与合成气(2015年8期)2015-12-23 10:40:22
能源(2015年8期)2015-05-26 09:15:44
中国酿造(2014年9期)2014-03-11 20:21:03
食品工业科技(2014年9期)2014-03-11 18:15:28
