
年青性染色体及鉴定方法
2022-07-28 08:26楚张卿罗玮夏云
四川动物 2022年4期
楚张卿 ,罗玮 ,夏云
(1.中国科学院成都生物研究所,成都 610041;2.中国科学院大学,北京 100049;3.绵阳师范学院生态安全与保护四川省重点实验室,四川 绵阳 621000)
脊椎动物的性别决定机制一般可以分为2种,即遗传性别决定和环境性别决定(Marshall,2008)。哺乳动物和鸟类的性别是遗传性别决定型且性染色体对高度异形化,如哺乳动物的性别决定系统是XX/XY型,即雄性异配,该系统中Y染色体高度退化;而鸟类的性别决定系统为ZZ/ZW型,即雌性异配,且W染色体高度退化。不同于哺乳动物和鸟类中高度异形化的性染色体,冷血脊椎动物鱼类、两栖类以及爬行类中大部分物种的性染色体在形态上既无太大差异,也未发生显著的分化(Hillis&Green,1990;Schmid&Steinlein,2001;Devlin&Nagahama,2002;Eggert,2004)。这种分化程度较低的性染色体被称为“年青性染色体”,即在性染色体分化后相当长的一段时间里,染色体形态及基因容量并无明显变化。
经典的性染色体分化模型认为,性染色体通常是由常染色体进化而来,常染色体对中的1条染色体因突变在偶然间获得了性别决定基因,如、或等(Charlesworth.,2005;Bachtrog.,2011;Bellott.,2014),该常染色体对就具备了一定的性别决定功能,被称为原始的性染色体(Beukeboom&Perrin,2014),该基因及其周围的部分区域之间的重组开始受到抑制,即重组抑制,以此来保护原始性染色体上的性别决定区域(Graves,2006)。随后性别拮抗基因在性别决定区域周围聚集,导致重组抑制区域沿着Y/W染色体逐渐扩大(Charlesworth.,2005),同时丢弃大量的无功能基因,并最终演变为形态上高度分化的异态形性染色体对(Bachtrog,2006,2013),如哺乳动物和鸟类中常见的高度异形化的Y或W染色体。因此重组抑制是性染色体分化的第一个阶段,也是最关键的一个阶段(Rice,1987),并随着其周围的性别拮抗基因和性别决定基因遗传给下一代。
经典的性染色体分化模型是从模式生物中的稳定且高度分化的性染色体对形成的模式提出的,如哺乳动物、果蝇等,但与哺乳动物和鸟类中高度分化的性染色体相比,鱼类、两栖类以及爬行类中大部分物种的性染色体的形态差异不大(Hillis&Green,1990;Schmid&Steinlein,2001;Devlin&Nagahama,2002;Eggert,2004)。而且性别决定系统或性染色体在这些类群中的不同科、属之间也存在差异,甚至在同一物种的不同地理种群中也不同(Ogata.,2003;Tanaka.,2007)。虽然有关鱼类、两栖类以及爬行类性染色体和性别决定的研究已经很多,但大多都集中于性染色体容易辨别的类群。由于年青性染色体在形态上难以识别,拥有这一类性染色体的非模式物种的性别决定和性染色体研究仍有许多未解之谜,但随着技术的发展(如高通量测序技术),关于年青性染色体研究也逐渐成为性染色体研究的热点,为性染色体进化提供了更全面的材料。
1 年青性染色体
经典性染色体进化假说,从提出到现在一直统治着整个性染色体进化的理论研究,该假说的确在少数特化类群中得到了很好的验证,如鸟类和哺乳动物,同时也引发了有关“性染色体生来就是要被毁灭的”世纪大讨论(Aitken&Marshall,2002;Steinemann&Steinemann,2005)。但随着染色体研究技术的不断突破以及对大量非模式物种的研究,研究者发现绝大多数具有年青同形性染色体(Vicoso&Bachtrog,2013)的物种中有大量未发生退化的同形性染色体。例如,鱼类中90%物种的性染色体(Devlin&Nagahama,2002)、两栖类中96%物种的性染色体为同形染色体(Hillis&Green,1990;Schmid&Steinlein,2001;Eggert,2004)。目前,对于这种年青性染色体普遍存在现象的解释,主要有性染色体“高频转换”假说和“不老泉”假说。比较基因组研究表明,性染色体在基因组中的位置往往是不稳定的,且经常在染色体之间发生转换(Bachtrog.,2014)。因而,推测同形性染色体之间的高频转换才是性染色体进化的常态,而当性染色体对之间的分化偶然超过某个阈值时,会掉入进化陷阱,这种情形下,性染色体的转换将被终止,性染色体继续退化而最终异形化(图 1)(Abbott,2017;Vicoso,2019)。因此,有学者提出,性染色体长期处于一种快速的新旧替换状态,没有足够的时间走向退化,从而维持其形态不变,保持着不断更新的“年青”状态(Volff.,2007;Vicoso,2019)。对多个支系进行的比较基因组学研究显示,一些演化支系的确已经发生了频繁的性染色体转换事件,包括两栖类(Hillis&Green,1990;Jeffries.,2018)、爬 行 类(Gamble.,2015)和鱼类(Myosho.,2015)。Gamble等(2015)在12种壁虎中发现了17~24次的性别决定系统的转化。Hillis和Green(1990)通过对63种蛙和蝾螈的性别决定系统类型分析,揭示了在两栖动物系统发育史上,性别发育系统从祖先的雌性异配型系统转换为雄性异配型系统,至少发生了8次转换。Jeffries等(2018)对蛙科Ranidae中的28个物种性别决定系统的研究,发现了至少13次性别决定系统转换事件,其中11次是XY与XY之间的转换。Roco等(2015)在热带爪蟾中曾发现了至少3种性染色体,且第三种性染色体处于过渡形态,存在于2个性别决定系统之间(Schartl,2015)。Miura等(2016)观察到,转换现象在粗皮蛙中也曾多次发生,因为在该物种中,同时存在XX/XY和ZZ/ZW 2种性别决定系统。
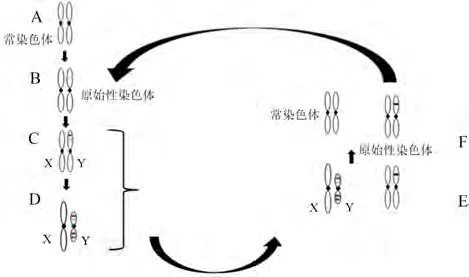
图1 性染色体形成的主流理论模型及性染色体转换(参考Abbott等,2017)Fig.1 The classical theoretical model of sex chromosome formation and sex chromosome transitions(referring to Abbott et al.,2017)
另一种假说是由Perrin(2009)提出的“不老泉”假说,该假说认为性反转个体中性染色体的偶然重组事件,维持着性染色体的同形状态。即使性染色体没有在异配性别中重组(如XY雄性),它们也可能在性反转个体(如XY雌性)中重组,因为重组模式取决于表型而不是基因型性别(Kondo.,2001;Lynn.,2005;Campos-Ramos.,2009;Matsuba.,2010)。Stöck等(2011)对3种近缘的欧洲雨蛙(、、)的研究发现,这3个物种都具有相同的同态形性染色体对,在雄性中完全不存在X-Y重组。尽管如此,X染色体和Y染色体之间的性别连锁位点序列并没有显示出差异。在系统发育分析中,X和Y等位基因是根据物种聚集的,而不是按配子进行聚类。因此推测这些树蛙的性染色体同态不是高频转换造成的,而是通过偶然的X-Y重组保持性染色体形态上的稳定。Stöck等(2013)在4种蟾蜍(、、、)中也发现了相同的性染色体偶然重组模式。
2 年青性别决定系统及性染色体的鉴定
对于性染色体分化程度高的物种来说,可以通过常规的核型分析鉴定性染色体和性别决定系统(李树深,胡健生,1999)。核型上的差异包括染色体数目差异(如XO)、染色体长度的差异、着丝粒位置差异、染色体长臂和短臂比值差异、甚至次缢痕差异等。与哺乳动物高度异化的性染色体不同,绝大多数的鱼类和两栖类存在年青性染色体,其在形态上的分化程度很低(即同形的性染色体)(Miura,2017),传统的细胞遗传学方法难以从核型形态上进行区分,仅部分可以通过G-带、C-带、Ag-NORs以及R-带等带型识别。这些带型反映了性染色体分化的程度,在部分物种中呈现两性差异,从而鉴定出同形性染色体(王子淑等,1983;李树深,胡健生,1996;张加一,谷晓明,1997)。但基于细胞遗传学的带型制作过程繁琐,结果准确率不高。
除了传统的带型分析以外,由于在性别决定区域存在序列差异,以及雌雄之间在基因表达层面也存在差异,多种基于高通量测序的方法也可用于判断性别决定系统和性染色体,分别是(1)基于DNA/RNA序列差异;(2)基于RNA表达差异;(3)基于蛋白质表达产物差异以及基于蛋白质组和代谢组之间的差异进行筛选区分。这些新兴的技术并不基于性染色体是否异形,大大提高了鉴定年青性染色体的效率。
2.1 比较DNA/RNA分子序列方法
高通量测序技术,包括DNA和RNA测序,已被大量用于性别连锁分子标记筛选和性别决定系统的鉴定。目前运用该技术已开发出大量的特异性分子标记,进行性别决定系统及性染色体的鉴定(Palmer.,2019)。
2.1.1 基于分子标记 通过筛选含有多态性的微卫星位点,再将多个雌雄个体进行等位基因分型,若雌雄含有不同的等位基因,则为性别连锁位点。在对罗非鱼的研究中,发现了6个与性别相关的微卫星位点,可用于性别的鉴定(陈文伟等,2020)。在棘腹蛙中,通过微卫星探针技术辨别染色体,不仅取得了良好效果,也为遗传多态性的研究奠定了基础(常晓嫒等,2013)。Yuan等(2017)利用1个性别特异性微卫星标记寻找棘腹蛙中的雄性异配子的同源模式,从而有助于解释性别决定系统和性染色体的进化。
扩增片段长度多态性(amplified fragment length polymorphism,AFLP)是一种检测DNA多态性的方法,通过限制性内切酶将基因组分解为大小不同的DNA片段,再对其进行PCR扩增,如在雌雄中出现不同的多态性,则为性别连锁标记。在研究香鱼养殖群体的遗传多样性时,通过AFLP标记技术对其进行性别特异性分子标记的筛选,最终筛选出1条雄性特异性分子标记的序列(闫松松等,2014)。刘雪清等(2015)尝试在成熟期长,且难以从外部形态鉴定性别的中华鲟基因组中寻找性别特异性的AFLP分子标记,虽然最后结果并不理想,但这些数据仍为中华鲟性别相关问题的研究奠定了基础。
单核苷酸多态性(single nucleotide polymorphism,SNP)及插入缺失(In/Del)分子标记具有独特的优势,包括密度高、遗传稳定等。且雌雄个体会固定不同的SNP及In/Del,因此可被用于性别决定系统及性染色体的鉴定。在对中国家养双峰驼进行研究时,通过在群体间进行Y-SNP多态标记的筛选,发现筛选出的Y染色体特异引物可以用于性别鉴定(陈慧玲,2016)。林晓煜等(2018)曾在大黄鱼中发现1个雄性特异SNP标记,并以此为基础开发出1种大黄鱼的新型遗传性别鉴定技术。Sun等(2018)针对黄姑鱼的基因在X和Y染色体上存在的差异设计出特异引物,以此对野生群体和养殖群体的遗传性别进行了鉴定。
2.1.2 基于关联分析 高通量测序出现后,基于关联分析可大批量筛选性别连锁的分子标记,主要测序分析方法包括全基因组关联分析(genomewide association study,GWAS)、限制性位点相关DNA测序(restriction site-associated DNA sequencing,RAD-seq)、基 因 分 型 测 序(genotyping-bysequencing,GBS)以及雌雄分库测序等,这些方法都是针对未退化完全的性连锁区域的序列,通常利用序列在性别间的关联情况来找出与性别关联的分子序列,并由此开发了RADSex等筛选软件(Feron.,2021)。基于关联分析的方法将雌雄分为2组,分析雌雄组间特有的差异,这种差异可以在SNP、K-mers、Reads等层面来反映。
Lambert等(2016)在林蛙中通过多样性阵列技术鉴定出8个雄性特有性别连锁SNP,表明该物种为XX/XY性别决定系统。Hu等(2019)为进一步了解大鲵的性别决定机制,利用限制性位点相关DNA(RAD)测序技术,分离出1个性别特异性遗传标记。类似于RAD-seq的方法,从DNA测序的数据中寻找性别特异的K-mers也可以鉴别出性别关联的序列。Duminda等(2020)基于K-mer值对澳大利亚三线石龙子的性别连锁序列进行鉴定,共鉴定出7个可靠的Y染色体特异性标记,表明其为XX/XY性别决定系统。Lin等(2020)在大黄鱼群体中进行了性别测定的GWAS,最终在18号染色体中确定了1个与雄性显著关联的SNP,为大黄鱼性别决定机制的确定奠定了基础。
在对大量样本进行测序时,为了降低测序成本、提高效率,出现了一种新型测序技术——Poolseq,其测序文库不是由单个个体或细胞的DNA制备的,而是由来自不同个体或细胞的DNA片段的混合物制备的(Schlötterer.,2014)。这种方法在鱼类性别研究中得到了大量应用(Wang T.,2019;Wen.,2019;Feron.,2020)。Zhang等(2017)在草鱼中利用Pool-seq鉴定了5个Y-连接的scaffold(总计347 kb)、6个Y-特异性序列和14个Y连锁基因,证明了基于新一代测序技术的DNA重测序技术在鱼类性别标记鉴定中的适用性,既为草鱼Y染色体的研究奠定了基础,也有助于阐明鲤科Cyprinidae鱼类性染色体的进化模式。
2.1.3 基于测序覆盖深度 针对已有基因组测序的物种,采集雌雄个体进行高通量测序或重测序,比较性染色体测序片段的深度可以判断性别连锁的区域和基因片段。XY系统中常染色体及雌性的2个X染色体之间有2个拷贝,而雄性的X染色体和Y染色体都只有1个拷贝。对基因组测序时,测序深度通常会是基因组大小的数十倍到数百倍。因此,从Reads的测序深度来说,如果一个区段雌性比对上的Reads比例是雄性的2倍,可确定该区域为性别连锁。该方法最早被用在了一些蛇类和鸟类的性染色体的确认和判断中(Vicoso&Bachtrog,2011;Zhou.,2014)。然而,这种方法仅适用于已经分化了足够时间的Y/W染色体,这是因为重组抑制积累了大量的变异,使得处于Y或W染色体上的序列不会被比对到X或Z染色体上(Charlesworth.,2021)。新近分化的性染色体类群分化程度较弱,性染色体(X与Y,Z与W)之间仍然具有很高的相似性,测序的Reads不能特异性地比对到其中一条性染色体上,从而使该方法失效。
2.1.4 基于哈迪-温伯格平衡 利用性染色体形成时重组抑制的特性,还可以利用哈迪-温伯格平衡的原理从种群遗传的角度来鉴定性别决定系统或性染色体。Käfer等(2021)基于自然种群中已知雌雄个体的等位基因和基因型的频率,利用启发式的概率计算方法(贝叶斯法),通过后验概率来检测性别连锁的基因。该方法比GWAS等方法所需的个体数少,每种性别的个体仅需5~10个。这种方法也可以使用RNA-seq、基因组重测序以及RAD-seq等的数据,其检测效率取决于染色体重组抑制区域的大小和分化程度。由于该方法最基本的原理是基于哈迪-温伯格平衡,因此理想样本是来自于同一大种群下的随机交配的个体,使用不同种群的标本可能会增加假阳性的风险。
2.1.5 基于遗传图谱 遗传图谱又称遗传连锁图谱,是指基因以及专一的多态性标记之间在基因组中相对位置的图谱,反映了染色体的交换与重组。早期遗传图谱的构建大多是利用显性和共显性标记方法,如AFLP和SSR标记,但随着下一代测序技术,如RAD-seq、GBS等方法的发展,以SNP为主要的遗传标记可构建更精细的遗传图谱,且根据此类遗传图谱还可以进行数量性状位点(quantitative trait locus,QTL)的检测(Gonen.,2014;Uchino.,2018)。因此,通过构建遗传图谱可以确定与性别性状有关的QTL位点(Gao.,2020),在大比目鱼遗传图谱的辅助下定位了其性别控制位点(Palaiokostas.,2013)。Qiu等(2018)构建了1个有8 094个SNP标记的黄姑鱼高密度遗传连锁图谱,并据此定位了QTL基因座以及相应区间,在QTL区间中鉴定并发现了124个性别二型性的候选基因,包括、和。Gao等(2020)利用 GBS构建了1个有5 705个SNP标记的黄颡鱼高密度遗传连锁图谱,并将其定位到26个不同的连锁群,鉴定发现出了11个显著的性相关QTL基因座,且在QTL区间内鉴定出6个性别相关基因,研究不仅阐明了黄颡鱼的性别分化过程,且为之后的分子辅助育种奠定了研究基础。
2.1.6 基于序列建树 这是一种通过基因树鉴定性染色体的新方法,即直接利用DNA/RNA鉴定。将测得的多个雌雄序列数据分别比对到参考基因组上,并在每条染色体上选取一定大小的窗口片段,将所有雌雄个体的2种分型都放在一起建树。如果来自所有雄性的一种分型全聚在一起,并且形成单系,那该基因树所在的片段是性别关联的片段,且反映了XY的性别决定系统。如果所有雌性的一种分型序列全聚为一支,则为ZW型性别决定系统。该方法首先需要构建X和Y序列的基因树,由于X和Y序列之间差异较大,理想状态下,X和Y序列分别是独立的2个树,由于序列很多,构建出的基因树也很多,此时哪棵树用分子标记可以把X和Y 2个基因树靠拢在一起,即为性别连锁标记,区别不开则不是(Dixon.,2019;Toups.,2019)。
2.2 基于RNA表达
利用两性之间一些基因在RNA表达水平上的差异,进行性别决定与性染色体的相关研究。Shen等(2020)基于RNA-seq分析筛选到鲇11个性别特异表达的基因,并使用qRT-PCR技术对11个基因进行验证,最终确定了8个性别特异表达基因。姚汶励等(2019)通过转录组测序的方法在草鱼雌雄样本中找到了数千个差异表达基因,最终发现基因和、基因分别与草鱼早期精巢和卵巢发育调控相关。Qin等(2020)对黄姑鱼性腺组织进行转录组测序,并基于基因表达差异筛选出了与性别相关的基因,即仅在卵巢中表达,而仅在精巢中表达。
2.3 基于蛋白质组学和代谢组学
在20世纪80年代蛋白质组学出现之前,学者普遍进行传统的蛋白质研究,即通过比较两性之间蛋白质的表达差异,从而确定性别决定系统,如同工酶法。Richard(1983)在中发现了一种性连锁酶——乌头酸酶,由于雌雄个体均携带这种酶,且乌头酸酶1号位点与性别决定基因连锁,便指出这些蛙类的性染色体可能表现为1对常染色体,且雄性性别决定基因位于其中1条同源染色体上。因传统的同工酶法筛选效率低、准确率不高,基于蛋白质组学和代谢组学可用于性别决定系统和性染色体的研究。
蛋白质组学提供了对特定细胞、组织或整个个体中所有受调控蛋白质的完整分析(Zhu.,2018),其可以通过研究蛋白质之间的相互作用以及蛋白质的表达修饰方式,来揭示蛋白质在生命过程中的作用(Wilkins.,1996)。代谢组学也是一门新的技术,研究目标是代谢小分子产物,如糖、有机酸和氨基酸等(Oliver,2002)。该技术有2个显著优势:一是在蛋白质水平或基因水平这类微小的变化可在代谢水平放大,进而更容易被观测到(Miracle&Ankley,2005;CuberoLeon.,2012);二是代谢产物的种类比基因或蛋白质的种类少,因此操作简便,无需再进行全基因组测序或建立表达序列的大型数据库的步骤(Wang L.,2019)。目前利用“组学”的方法进行科学研究越来越成熟,可以尝试运用这类“组学”方法对性染色体分化程度较低物种的性别决定机制进行研究,Wang T等(2019)采用了蛋白质组和代谢组相结合的方法,发现在河川沙塘鳢性别决定中起重要作用的蛋白质有4种,分别是 Ctnnb1、Piwil1、Hsd17b1和 Dnali1,而在性别决定中起重要作用的代谢过程是脂代谢。在上述实验中,通过使用定量PCR技术以及原位杂交技术,发现了目标蛋白质在雌雄个体中的表达不同,为进一步明确河川沙塘鳢的性别决定机制奠定了基础。
3 展望
由于年青性染色体系统的类群演化模式多种多样,甚至于同一物种或近缘种的性别决定机制或性染色体都会有所不同,并且与性别相关联的区域相对于基因组来说太小,因此需结合多种鉴定技术,从而准确鉴定性别决定系统和性染色体。在未来的研究中,可以将测序技术、组学数据和生物信息学结合起来,对非模式物种的性别决定系统及基因组进行深入研究,而这种综合研究可以算作性别组学的研究起点。对于新出现的概念——性别组学,几乎涵盖了基因组、转录组以及蛋白质组的全部信息(Stöck.,2021)。这种综合性的组学方法将开启有关性染色体研究的新时代,它可以深入阐明脊椎动物的性别决定及性染色体进化问题,揭示其背后的遗传机理,也可解释更多年青性别决定系统的问题。到目前为止,在变温脊椎动物中,有关性别决定系统与生殖模式共同进化的信息还知之甚少(Charnov&Bull,1977;Warner&Shine,2008;Organ.,2009)。在此研究背景之下,未来的测序方向应集中在缺乏性别决定信息和生殖模式信息的物种上(Stöck.,2021)。利用性别组学的方法,将会对非模式脊椎动物的性染色体序列信息有更深入的理解,也将会最终阐明性别决定为何有如此多的方式以及性别决定系统进化的动力和分子途径。
感谢中国科学院成都生物研究所两栖爬行类动物研究室的曾晓茂研究员和郑渝池副研究员在文章题材选取和内容讨论以及文稿撰写过程中给予了宝贵意见。
猜你喜欢
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
波谱学杂志(2022年1期)2022-03-15
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
分析化学(2019年3期)2019-03-30
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
分析化学(2017年12期)2017-12-25
小学教学参考(语文)(2017年11期)2017-11-30
