
长链非编码RNA在动脉粥样硬化“损伤-应答”中的作用机制研究进展
2022-07-14 03:04陈宇恒王正龙
解放军医学杂志 2022年6期
陈宇恒,王正龙
遵义医科大学附属医院心血管内科,贵州遵义 563000
动脉粥样硬化(atherosclerosis,AS)的发生发展机制可简要概括为“损伤-应答”学说,即多因素导致血管内皮细胞(vascular endothelia cell,VEC)功能失调,单核-巨噬细胞(Mo-Mφ)及淋巴细胞介导动脉内膜炎症,导致血管平滑肌细胞(vascular smooth muscle cell,VSMC)迁移增殖及泡沫细胞沉积,最终以斑块破裂、钙化或纤维化为结局[1-3]。尽管目前针对AS的治疗策略已明显改善了患者的预后,但长期用药、药物不耐受及用药禁忌等问题的存在,使AS的合理、规范化治疗仍面临一定挑战[4-5]。长链非编码RNA(long non-coding RNA,lncRNA)是一类核酸序列长度大于200 bp的非编码RNA,因其缺乏开放阅读框,不通过编码蛋白调控生物体机能[6],曾被认为是人类基因组中的“噪点”及“暗物质”。随着高通量测序与蛋白组学的发展,人们发现lncRNA可在转录、翻译、翻译后修饰等多个层面影响疾病的发生发展过程[7-10],且目前已有lncRNA参与“损伤-应答”调控流程的相关报道。因此,本文汇总了NEXN-AS1、MANTIS、LeXis、MALAT1等lncRNA调控“损伤-应答”的机制,旨在为进一步明确AS的发病机制、完善AS的诊疗提供参考。
1 LncRNA NEXN-AS1的抗炎效应与“损伤-应答”
1.1 NEXN与心血管病 NEXN(nexilin-F-actinbinding-protein)在心肌细胞中作为T管的核心,与Jph2(junctional protein junctophilin 2)、RyR2(type 2 reynoldin receptor)共定位于心肌肌质网,组成膜连接复合体以调控心肌细胞膜的钙电流及胞质内的钙瞬变[11],但在VSMC、VEC、Mo-Mφ细胞中则作为肌丝结合蛋白,并调节细胞的黏附及迁移[12]。近年来研究发现,NEXN在人、鼠等多个物种间高度保守,且与多种心血管疾病存在较强的关联,在小鼠模型中,NEXN已被证实参与了AS、扩张型心肌病的病理生理过程[11,13];一项大规模的多中心随机对照临床试验也提示NEXN参与了部分扩张型心肌病的进展[14];此外,冠心病患者外周血、病灶中NEXN的表达水平也低于健康人[12]。
1.2 LncRNA NEXN-AS1调控“损伤-应答”的机制 2019年,Hu等[12]报道,NEXN-AS1(NEXN antisense RNA 1)作为NEXN的反义RNA与NEXN共定位于1p31.1。
Hu等[12]通过高脂喂养ApoE-/-小鼠模拟脂代谢紊乱,通过LPS处理VEC/VSMC/Mφ模拟血管内炎症,并基于此探讨NEXN-AS1对动脉粥样硬化“损伤-应答”的调控机制,发现NEXN-AS1的5'端1~1000 nt序列可与组蛋白的重塑剂——溴基结构域相邻的锌指结构域蛋白1A (bromodomain adjacent to zinc finger domain 1A,BAZ1A)交联,进而使染色质由致密变得疏松,上调NEXN的表达。此外,该研究还发现,作为NEXN-AS1在表观遗传性状调控上的映射,VEC的Toll样受体4/核因子κB(Toll like receptor-4/nucler factor-κB,TLR-4/NF-κB)通路被下调,单核细胞趋化蛋白-1(monocyte chemoattractant protein-1,MCP-1)、肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白细胞介素-6(interleukins-6,IL-6)、细胞黏附分子-1(intercellular adhesion molecule 1,ICAM-1)、血管黏附分子-1(vascular adhesion molecule 1,VCAM-1)的表达水平降低,最终,Mo-Mφ的趋化及VSMC的增殖被抑制,炎症反应对VEC的损伤作用减轻。总之,NEXN-AS1的下游效应几乎涉及了“损伤-应答”反应的每一个阶段,详细过程见图1。
1.3 NEXN-AS1/NEXN与细胞焦亡 细胞焦亡(pyroptosis)是有别于细胞凋亡、自噬性死亡的一种程序性死亡方式,以半胱氨酸天冬氨酸蛋白酶1(caspase-1)、消皮素D(gasdermin D,GSDMD)、NOD样受体热蛋白结构域相关蛋白3(NOD-like receptor thermal protein domain associated protein 3,NLRP3)、白细胞介素-1β(interleukin 1β,IL-1β)作为分子标志[15],几乎涉及“损伤-应答”的各个环节:VEC焦亡促进了Mo-Mφ在血管内膜的募集,巨噬细胞焦亡加速了AS坏死核心的形成[16],VSMC焦亡则削弱了斑块纤维帽的厚度,可促进不稳定斑块的形成[17]。
Wu 等[18]使用不同浓度的阿托伐他汀处理VEC,发现NEXN-AS1/NEXN的表达呈剂量依赖方式增加,而细胞焦亡的标志分子caspase-1、GSDMD、NLRP3、IL-1β等则被抑制,NEXNAS1/NEXN被沉默后,阿托伐他汀的抗焦亡效应消失,提示他汀的内皮保护作用与lncRNA相关,为lncRNA作为AS的干预靶点提供了证据。
1.4 NEXN-AS1/NEXN的应用前景 有研究采用基因微阵列分析正常及AS病变的主动脉组织,结果发现NEXN-AS1/NEXN在粥样斑块中的表达水平与病变程度呈负相关[12]。此外,人VEC NEXNAS1/NEXN沉默后,caspase-1、GSDMD、NLRP3、IL-1β的表达明显增高,且小鼠体内NEXN-AS1被沉默后,可明显加速AS的进展[18]。就机制来说,NEXN-AS1广泛参与了血管内炎症及细胞焦亡的调控(图1),就表观性状来说,NEXN-AS1所调控的NEXN涉及AS在内的多种心血管疾病,提示NEXNAS1或可成为治疗疑难心血管疾病的新靶点。
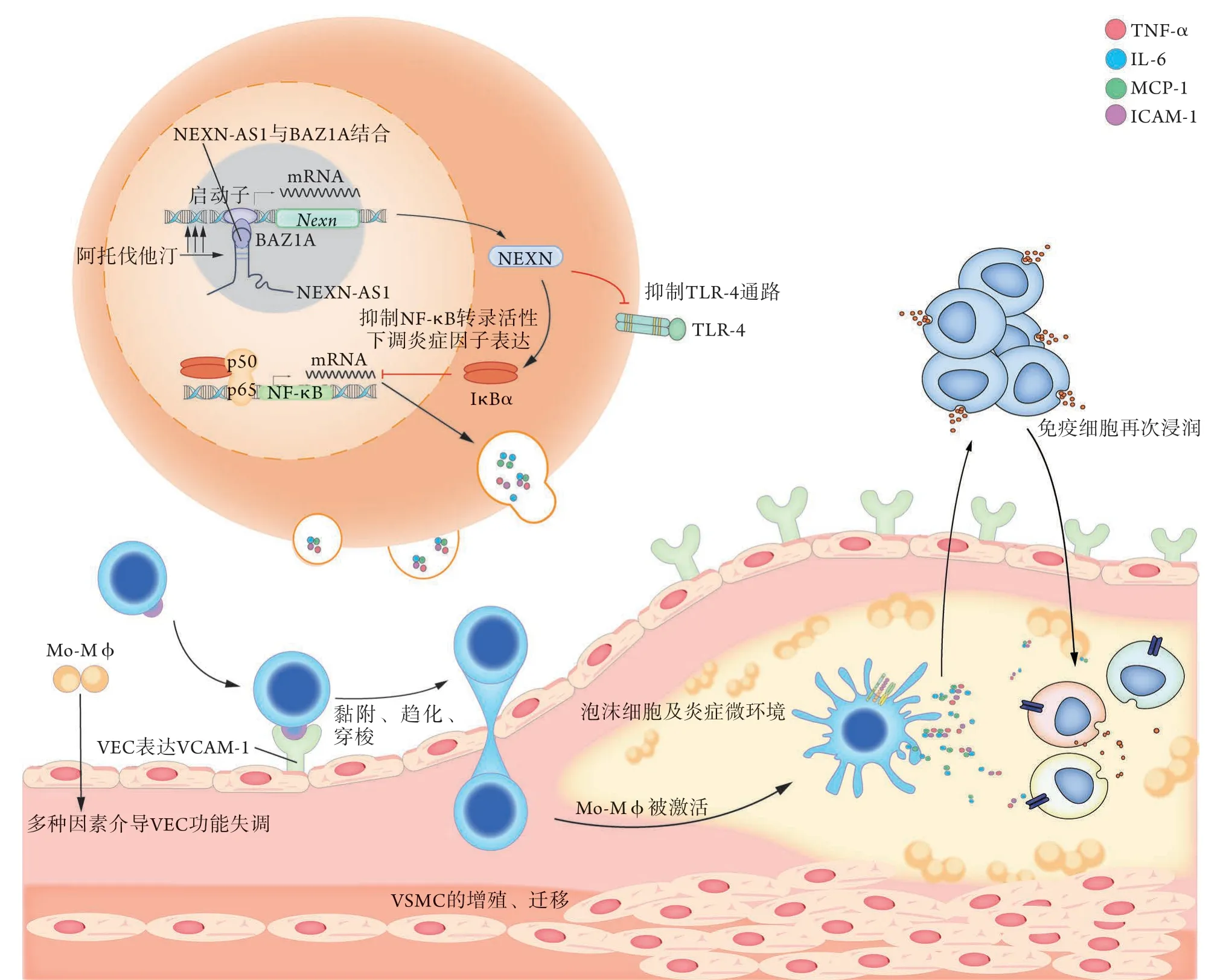
图1 NEXN-AS1通过NENX抑制“损伤-应答”的机制Fig.1 NEXN-AS1 inhibits the progression of "Injury-Response"
2 LncRNA MANTIS的他汀样作用与“损伤-应答”
2.1 LncRNA MANTIS概述 2017年,Leisegang等[19]报道了lncRNA MANTIS,其基因位于2p13.3,为Anxa4的反义内含子,在人与小鼠间高度保守,与VEC的增殖、分化密切相关。MANTIS受H3K4赖氨酸特异性去甲基化酶5(JARID1B)、肌肉细胞特异性增强因子2A(myocte specific-enhancer factor-2A,MEF2A)的负向调控及Krüppel样转录因子2(KLF2)/KLF4、他汀类药物的正向调控,广泛表达于各种内皮细胞[19-20];此外,在食蟹猴的斑块消退期可检测到MANTIS的高表达,而在人颈动脉粥样斑块内MANTIS则呈低表达[19]。
2.2 LncRNA MANTIS与SOX18/SMAD6/COUP-TFⅡ在血管内皮生长、分化中的作用 目前认为,性别决定区Y框蛋白18(sex determining region Y-box 18,SOX18)、Smad同源物6(SMAD6)、鸡卵清蛋白上游启动子转录因子Ⅱ(chicken ovalbumin upstream promoter transcription factor Ⅱ,COUP-TF Ⅱ)在VEC的生长、分化过程中起着至关重要的作用[21-23]。当敲除MANTIS后,SOX18、SMAD6、COUP-TF Ⅱ的表达下调,VEC的出芽、增殖、迁移能力受阻;此外,MANTIS不仅促进了SOX18、SMAD6、COUPTF Ⅱ的表达,还促进了三者间的功能整合[19]。
MANTIS的外显子3为一段含Alu元件的序列,对SOX18、SMAD6、COUP-TF Ⅱ的功能整合及稳定染色质重构复合物核心催化亚基(brahma related gene-1,BRG-1)/BAF155复合物至关重要:在球体生长试验中,敲除VEC的MANTIS后,单纯过表达SOX18、SMAD6、COUP-TFⅡ不能使内皮的发芽正常化,而过表达Alu元件则可抵消MANTIS敲除的效应,提示MANTIS的生物学功能依赖于其含Alu元件的序列[19]。此外,MANTIS的Alu元件还可通过抑制ICAM-1的表达来抑制Mo-Mφ在血管内膜的黏附、激活,提示其或可抑制由各种“损伤”引起的Mo-Mφ“应答”[20]。
2.3 LncRNA MANTIS通过SWI/SNF调控内皮功能的机制 SWI/SNF复合物(SWItch/sucrose nonfermentable complex)是广泛存在于哺乳动物细胞核中的ATP依赖的染色质重塑剂,依托乙酰化组蛋白H3K27定位于染色质[24];目前认为SWI/SNF复合物以BAF47/155/170(SWI/SNF complex 47/155/170 kD subunit)、BRG-1为分子核心[25],通过BRG-1提供ATP酶活性,以滑动、消除组蛋白的方式创建DNA区域,实现基因表达的调控[24,26]。
LncRNA MANTIS可稳定BRG-1/BAF155复合物,促进SWI/SNF复合物介导的染色质重塑,通过提高RNA聚合酶Ⅱ的转录活性来提高SOX18、SMAD6、COUP-TF Ⅱ的表达水平,促进VEC的增殖分化及功能整合[19](图2)。

图2 LncRNA MANTIS对内皮细胞生长、分化的促进作用Fig.2 Effect of lncRNA MANTIS promotes endothelial growth and differentiation
2.4 LncRNA MANTIS与他汀的多效性 尽管目前尚无lncRNA MANTIS直接参与AS发生发展的报道,但作为抗AS基石之一的他汀类药物可通过增强MANTIS启动子活性、诱导KLF2/KLF4、抑制MEF2A等多种途径调控MANTIS的表达[20],且在MANTIS敲除的内皮细胞中,阿托伐他汀的促血管再生、调控血栓调节蛋白转录谱、增加端粒酶活性等抗AS效应均被抑制,提示lncRNA MANTIS或可抑制AS进展,并成为抗AS治疗的潜在靶点[20]。
3 LncRNA LeXis的调脂效应与“损伤-应答”
3.1 LncRNA LeXis概述 LncRNA LeXis(liverexpressed LXR-induced sequence)又名CT70,基因定位于9p31.1,由Sallam等[27]于2017年在探究肝X受体(liver X receptor,LXR)、固醇调节元件结合蛋白(sterol regulatory element-binding protein,SREBP)调控胆固醇代谢的过程中发现并报道,命名为LeXis,其在人与小鼠间均高度保守。
3.2 LncRNA LeXis的调脂效应与SREBP SREBP本身作为调控胆固醇生物合成的重要分子,在机体固醇含量丰富时被抑制,反馈性下调胆固醇的生物合成途径[28-29]。LeXis通过促进胆固醇流出肝脏,并抑制胆固醇生物合成的方式调控胆固醇稳态,而LeXis的调脂效应不同于他汀、前蛋白转化酶枯草素/Kexin9(proprotein convertase subtilisin/kexin type 9,PCSK9)抑制剂的药理作用,不影响肝功能,无内质网应激,也无需低密度脂蛋白受体(LDL-R)的参与,提示LeXis可能干预脂代谢紊乱所引起的“损伤”,进而抑制下游的“应答”[4,27]。
Sallam等[27]于小鼠肝细胞过表达LeXis后,提取核蛋白行质谱分析时发现了含RNA结合结构域、亮氨酸拉链结构域的核糖核蛋白RALY,其本身是小鼠肝脏胆固醇生物合成基因的转录辅助因子(transcriptional cofactor)激活剂,而LeXis正是通过与RALY交联,减少RNA聚合酶Ⅱ在Srebp2等靶基因启动子上的起始转录,进而降低SREBP2、3-羟基-3-甲基戊二酸单酰辅酶A(HMG-CoA)还原酶、法尼基二磷酸法尼基转移酶1(FDFT1)的表达,最终使胆固醇的生物合成明显减少(图3)。
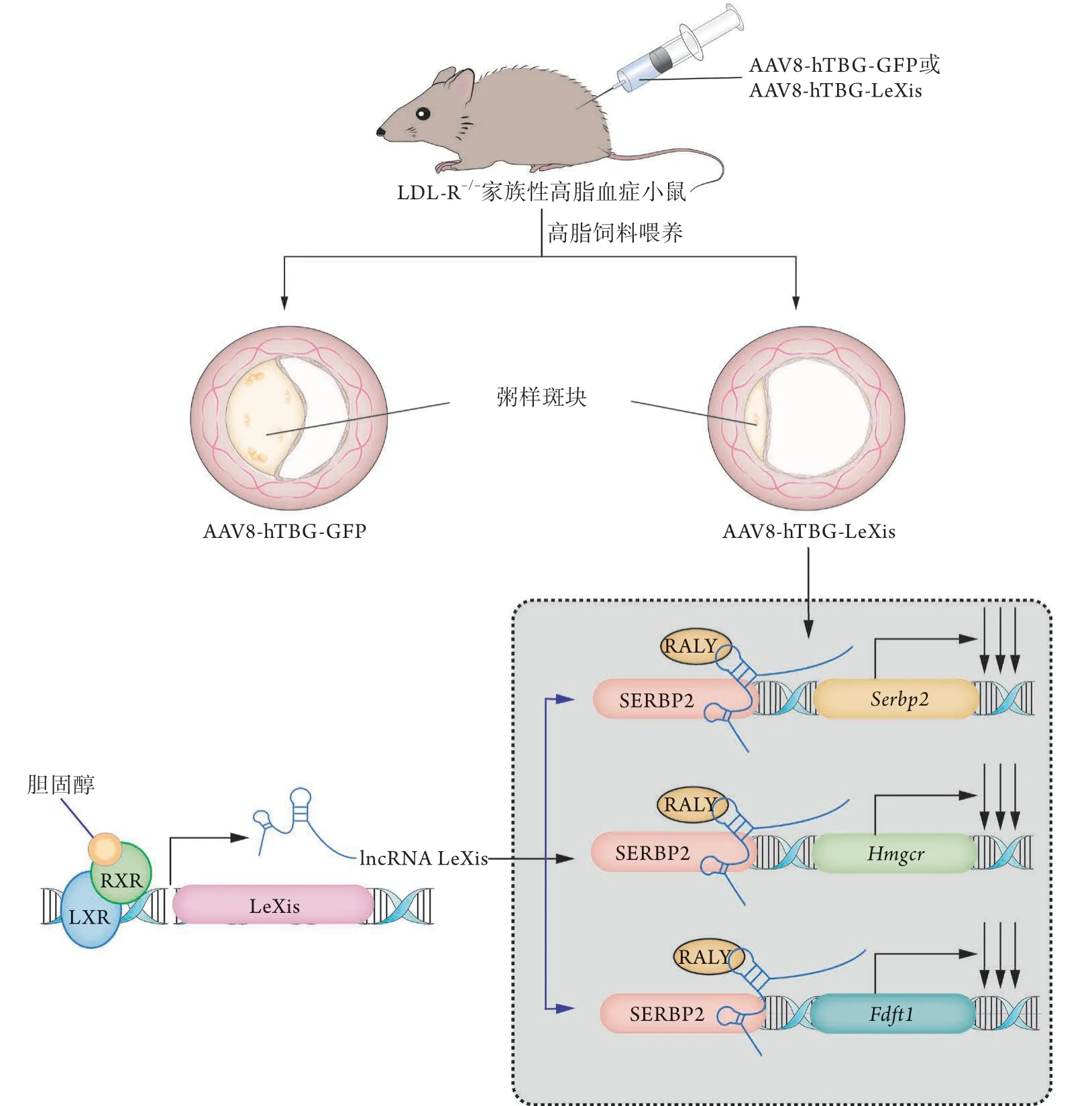
图3 LncRNA LeXis对脂代谢的调节机制Fig.3 Regulatory mechanism of lncRNA LeXis on lipid metabolism
3.3 LncRNA LeXis的应用前景 家族性高胆固醇血症(familial hypercholesterolemia,FH)是以血浆低密度脂蛋白胆固醇(LDL-C)明显升高为特征的一种常染色体遗传病,多数FH患者因涉及Ldlr、Apob的突变,即使高强度他汀、PCSK-9抑制剂治疗也无法控制LDL-C达到指南的推荐目标[4,30-31]。
Tontonoz等[31]将含LeXis的AAV-8质粒转染入LDL-R-/-FH小鼠,发现LeXis处理后的小鼠总胆固醇及三酰甘油水平明显降低,且AS病变程度明显减轻(图3),提示LeXis可用于治疗FH等难治性高脂血症,强化AS或冠心病的各级预防,改善此类患者的预后,也再次为lncRNA可作为治疗靶点提供了证据。
4 LncRNA MALAT1的免疫调控与“损伤-应答”
4.1 LncRNA MAL AT1概述 肺腺癌转移相关转录本1(metastasis-associated lung adenocarcinoma transcript 1,MALAT1)基因位于11q13.1,序列长8 kb,在人与小鼠间高度保守。初始MALAT1在其多聚腺苷酸上游可形成类似tRNA的三叶草结构,由RNA酶P (RNase P)识别、剪切形成两个转录本,其中的5'端侧即为7000 nt的成熟MALAT1,而3'端侧经RNase Z、CCA添加酶修饰后进入胞质,形成MALAT1-associated small cytoplasmic RNA(mascRNA)[32](图4)。
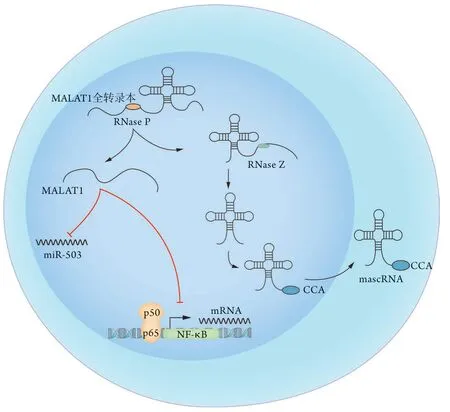
图4 LncRNA MALAT1的加工过程及下游靶分子Fig.4 Processing of the lncRNA MALAT1 and its downstream target molecules
4.2 LncRNA MAL AT1的免疫活性调节机制MALAT1通过与巨噬细胞、树突状细胞(dendrite cell,DC)中的NF-κB结合而沉默NF-κB,调节巨噬细胞与DC的天然免疫应答,调控血管内膜炎症[33]。T细胞在粥样斑块中占白细胞的25%~38%[34],提示T细胞在“损伤-应答”调控中占有重要地位,而MALAT1可通过抑制DC胞内的NF-κB与CD80启动子结合,下调DC膜表面CD80的表达[35],从而削弱初始T细胞的激活,进一步减弱各种“损伤”因素引起的Mo-Mφ、DC-T细胞“应答”。
M A L A T 1 的另一部分抗炎机制依赖于mascRNA:在MALAT1+/-骨髓源性巨噬细胞株(bone marrow macrophage,BMDM)的趋化、促炎症因子(TNF、NOS2、CCL2、CCL7)水平明显高于野生型BMDM的基础上,利用锁核酸修饰的反义寡核苷酸(lock nucleic acid modified antisense oligonucleotides,L N A-A S O)选择性耗竭M A L AT 1+/+B M D M s 的mascRNA后,其TNF、IL-6的表达水平可更进一步升高,提示mascRNA也参与了免疫炎症的调控,但目前尚缺乏其机制的研究报道[36]。
MALAT1还可通过miR-503的分子海绵效应发挥对“损伤-应答”的负调控作用:Cremer等[37]、Yan等[38]分析了MALAT1+/-小鼠与野生型小鼠的差异性microRNA,发现包括miR-503在内的多种microRNA在转录水平不变的背景下含量增加,而抗miR-503可显著降低TNF-α介导的VEC表型转换,模拟MALAT1的内皮保护效应。
4.3 LncRNA MALAT1与AS的关联 Gast等[36]发现,即使在正常饮食喂养下,MALAT1+/-ApoE-/-小鼠的AS进展也快于ApeE-/-小鼠。Cremer等[37]对MALAT1+/+ApoE-/-小鼠行MALAT1-/-ApoE-/-骨髓移植,发现可解除MALAT1的AS保护效应,粥样斑块也更趋向发展为不稳定斑块。有临床研究也发现粥样斑块内的总MALAT1水平或与患者心脑血管事件的发生率呈负相关,提示MALAT1可抑制AS的进展[36-37]。
5 其他参与调控“损伤-应答”的lncRNA
5.1 LncRNA MeXis通过ABCA1调控“损伤-应答”的机制 MeXis (macrophage-expressed LXR-induced sequence)由Sallam等[39]于2018年报道,在人与小鼠间高度保守,可被LXR激活,通过DDX17-ABCA1轴进行调脂,抑制脂代谢紊乱引起的“损伤”,进而抑制下游的“应答”。
MeXis可协助DDX17(一种核受体辅助因子)与LXR在Abca1启动子上结合,激活ATP结合盒转运元件A1(ATP binding cassette transporter A1,ABCA1)的表达,而ABCA1则通过促进游离胆固醇流向Apo A-1,加速HDL-C的生成,促进巨噬细胞内的脂质流出[40]。
在动物模型中,MeXis-/-LDL-R-/-小鼠无论是AS进展还是易损斑块的发生率均高于LDL-R-/-小鼠,且一项基于1000个基因组的冠脉疾病全基因组关联meta分析结果发现,人MeXis单核苷酸多态性与冠心病存在明显相关性,提示MeXis可抑制AS的进展[41]。
5.2 LncRNA SNGH-12促进DNA修复调控“损伤-应答”的机制 氧化应激可致DNA损伤,加速VEC的功能障碍,继而促进“损伤-应答”[42],小核仁宿主基因-12(small nucleolar host gene-12,SNHG-12)在进化上较为保守,可通过DNA依赖性蛋白激酶(DNAdependent protein kinase,DNA-PK)/DNA-PKcs-Ku70/Ku80途径,辅助VEC的DNA损伤修复[43]。
大多数哺乳动物以非同源末端连接(nonhomologous end-joining,NHEJ)的方式修复DNA双链断裂:通过Ku70/Ku80异二聚体快速识别断裂的DNA末端并保护其免受核酸酶水解,随后DNA-PK的催化亚基(DNA-dependent protein kinase catalytic subunit,DNA-PKcs)提供激酶活性,磷酸化大量与之重叠的底物,促进有效而准确的DNA修复,通过抑制“损伤”的进展,继而抑制下游的“应答”[44-46]。
LncRNA SNHG-12可与DNA-PK交联,促进DNA-PK/DNA-PKcs与Ku70/Ku80的相互作用,提高NHEJ的效率,抑制氧化应激,而敲除SNHG-12可加速VEC的衰老,促进AS进展[43]。
5.3 LncRNA MARRS抑制胞葬作用调节“损伤-应答”的机制 巨噬细胞相关动脉粥样硬化基因序列(macrophage-associated atherosclerosis lncRNA sequence,MARRS)由Simion等[47]于2020年报道,可通过调节HuR(一种RNA结合蛋白,可结合凋亡相关基因的mRNA,抑制细胞凋亡)而加速巨噬细胞的凋亡,抑制斑块内的胞葬作用,促进“损伤-应答”的进展。
胞葬作用是细胞在发生进一步坏死之前清除凋亡细胞的过程[48]。在斑块早期,VSMC及巨噬细胞的凋亡可被胞葬作用清除,但随着AS进展,巨噬细胞凋亡的累积使胞葬作用不足以维持正常的血管内膜结构;最终,斑块内坏死不断加重,斑块性质转变为易损斑块[49]。
LncRNA MARRS可通过沉默HuR,促进p53、p27、caspase-8和caspase-9表达,增加巨噬细胞的凋亡,从而削弱巨噬细胞的胞葬作用,加速AS的进展[47]。
5.4 其他调控“损伤-应答”的LncRNA
5.4.1 促进“损伤-应答”的lncRNA
5.4.1.1 LncRNA VINAS LncRNA VINAS (Vascular INflammation and Atherosclerosis lncRNA Sequence)与人DEPDC4(DEP domain containing 4)同源,在VEC/VSMC/Mo-Mφ中均有表达,但主要表达于VEC[50],可通过NF-κB和MAPK通路调节血管内炎症,敲低VINAS可减少VEC/VSMC/Mo-Mφ源的MCP-1、TNF-α、IL-1β水平,抑制AS的进展,且人DEPDC4的敲低也可复制VINAS的抗炎效应。
5.4.1.2 LncRNA MEG3 LncRNA MEG3(maternally expressed gene 3)在人、鼠间保守,并表达于多种组织[51];MEG3可充当miR-223(一种重要的抗炎miRNA[52])的分子海绵,以增加NLRP3、凋亡相关斑点样蛋白(apoptosis-associated speck- like protein containing a CARD,ASC)、cleaved caspase-1及GSDMD的表达,促进VEC的焦亡并加速AS进展[53],而具有抗AS效应的褪黑素则可阻断MEG3/miR-223/NLRP3轴以缓解MEG引起的VEC焦亡,提示lncRNA MEG3或可作为AS的干预靶点[53]。
5.4.1.3 LncRNA Kcnq1ot1 KCNQ1重叠转录本1(kcnq1 overlapping transcript 1,Kcnq1ot1)是Kcnq1基因座上的一种印记反义lncRNA[54],在人与小鼠间保守[55]。lncRNA kcnq1qt1通过竞争性结合miR-452-3p以增强组蛋白去乙酰化酶3(histone deacetylase 3,HDAC3)的表达,进而减少ABCA1的表达,使巨噬细胞的胆固醇流出受抑制,最终加重脂代谢紊乱;kcnq1qt1敲除则可抑制人单核细胞白血病细胞系(human monocytic leukemia cell line,THP1)源巨噬细胞的脂质积聚,并明显减缓ApoE-/-小鼠的AS进展[54]。
5.4.2 抑制“损伤-应答”的lncRNA
5.4.2.1 LncRNA NORAD DNA损伤激活的非编码RNA(non-coding RNA activated by DNA damage,NORAD)是一种在哺乳动物高度保守,并参与调节基因组稳定性的lncRNA[56],可通过抑制NF-κB、p53-p21及IL-8来调控细胞周期、血管内炎症,并发挥VEC的保护效应[57]。NORAD敲除后,ox-LDL诱导的活性氧、NF-κB及其下游的ICAM、VCAM、IL-8增加,加速了ApoE-/-小鼠的AS进展[57]。
5.4.2.2 LncRNA PEBP1P2 LncRNA PEBP1P2是VSMC表型转化的调节因子,不仅可直接与细胞周期依赖的蛋白激酶9(cyclin-dependent kinase 9,CDK9)结合,下调p38-MAPK通路的c-Jun、p38磷酸化水平以拮抗AS进展,还可抑制血小板衍生生长因子BB(PDGF-BB)诱导的VSMC表型转换[58]。基于PEBP1P2可直接结合CDK9而抑制VSMC增殖、迁移的特性,提示其可能成为晚期AS的治疗靶点。
5.4.2.3 LncRNA-FA2H-2 LncRNA-FA2H-2的抗AS效应主要依赖于混合谱系激酶结构域样蛋白(mixed lineage kinase domain-like protein,MLKL),后者可引起哺乳动物雷帕霉素靶蛋白(mechanistic target of rapamycin kinase,mTOR)/蛋白激酶B(protein kinase B)依赖的VEC/VSMC自噬缺陷,促进MCP-1、VCAM-1、IL-6的表达[59],而lncRNA-FA2H-2的-750~387序列可与MLKL的启动子区域结合,通过下调MLKL的表达而缓解自噬缺陷及炎症反应,减缓“损伤-应答”的进展[59]。其余参与“损伤-应答”调控的lncRNA见表1。

表1 促进及抑制“损伤-应答”的lncRNATab.1 LncRNAs that accelerated and inhibit the "Injury-Response" process
6 总结与展望
NEXN-AS1、MANTIS、LeXis、MALAT1等lncRNA高度保守,具备强大的表观遗传调控能力,并参与调控“损伤-应答”中的脂质沉积、血管内膜炎症、细胞增殖与凋亡等过程,因此,lncRNA可能成为AS新的诊疗靶点。但目前仍存在一些争议,如肿瘤相关lncRNA用于治疗是否会增高肿瘤发病率,lncRNA促进VEC增殖是否会加速支架置入术后的支架内再狭窄,以及lncRNA是否可改善AS的远期预后等。希望心血管领域的科研人员更加重视lncRNA的地位,早日解决上述问题,以实现lncRNA的临床应用,改变目前AS诊疗的窘境,最终使AS患者获益。
猜你喜欢
中国计划生育和妇产科(2022年6期)2022-11-15
世界科学技术-中医药现代化(2022年3期)2022-08-22
实用临床医药杂志(2022年13期)2022-07-26
传染病信息(2022年3期)2022-07-15
现代临床医学(2022年3期)2022-06-06
医学综述(2022年7期)2022-04-19
自我保健(2021年2期)2021-11-30
现代仪器与医疗(2021年4期)2021-11-05
妇女之友(2021年9期)2021-09-26
皮肤病与性病(2021年3期)2021-07-30
