
离子聚合物不同间隔单元对催化CO2环加成反应的影响
2022-07-12 07:40:02朱立华肖志音何亚兵
无机化学学报 2022年7期
朱立华 肖志音 钟 伟*, 何亚兵
(1浙江师范大学化学与生命科学学院,金华 321004)(2嘉兴学院生物与化学工程学院,嘉兴 314001)
0 引 言
二氧化碳的大量产生与排放导致了全球变暖、海平面上升等一系列严重的气候和环境问题。在这种现实背景下,世界主要经济体开始通过多项行政、经济手段,逐步加强对碳排放的控制。我国也于2020年9月宣布“CO2排放力争于2030年前达到峰值,努力争取2060年前实现碳中和”。2021年政府工作报告再次明确强调“扎实做好碳达峰、碳中和各项工作”。实现碳中和,一方面要在源头上减少碳排放,另一方面则要想方设法降低空气中的CO2浓度,变废为宝,把已排放的CO2作为资源加以利用。事实上,作为一种无毒、廉价且丰富的C1资源,CO2可以转化为多种高附加值的工业产品,如甲酸、甲烷、甲醇以及碳酸酯等。其中CO2与环氧化物转化生成环状碳酸酯的环加成反应具有良好原子经济性,近年来引起了人们的广泛关注。
迄今为止,已有大量催化CO2与环氧化物环加成反应制备环状碳酸酯的催化剂被研发和报道,包括均相催化剂如金属卤化物、离子液体(ILs)、有机碱和金属配合物[1-4],多相催化剂如金属氧化物[5]、聚离子液体(PILs)负载型催化剂[6-9]、金属有机骨架(MOFs)材料[10-13]和碳基催化材料[14-15]等。但均相催化剂不易回收循环、产物难分离,多相催化剂往往具有反应条件苛刻、催化剂易损失以及需要可溶性的共催化剂等缺点,均在一定程度上限制了其广泛应用。因此,发展多相催化剂,实现在温和、无共催化剂条件下的CO2环加成高效转化,成为该研究领域科研工作者们的努力方向。
另一方面,近年来发展起来的多孔离子聚合物具有合成简单、热稳定性好、结构功能“可设计性”强、高比表面积、较高的官能团含量以及高电荷密度等特征,成为催化领域中一种理想的候选材料,已引起了广泛的关注和研究[6-8,16-17]。由于多孔离子聚合物孔道内固有的离子位点有利于增强与气体分子间的相互作用,为吸附分子提供了有利的化学环境,其整体结构具有的高电荷密度、高极化力、高比表面积和强相互作用力,使得多孔离子聚合物不但对CO2气体具有很强的吸附能力,而且可作为CO2转化为高附加值化学产品的高效催化剂。事实上,已有不少关于多孔离子聚合物催化CO2与环氧化物环加成制备环状碳酸酯的报道,表现出良好的催化活性[6-8]。但这些催化剂大都需要在较高的反应温度或较大的CO2压力条件下才能实现高效转化,并且随着温度或压力的降低,环状碳酸酯的产率明显下降。因此,如何通过催化剂的合理设计以实现CO2在常温、常压温和条件下的高效环加成反应,仍然是一个很大的挑战。
我们分别以含有苯基、吡啶基、酚羟基的多卤化物和N-(三甲基硅基)咪唑作为前体,通过一锅直接季铵化法合成了3种具有不同间隔单元(spacer)的多孔离子聚合物IP1~IP3,考察了这类聚合物间隔单元的变换对催化CO2与环氧化物环加成制备环状碳酸酯的性能影响,并测试其催化循环及稳定性。研究结果表明:在无溶剂、80℃和pCO2=101 kPa的条件下,3种具有不同间隔单元的离子聚合物均可以超过93%的产率(反应时间12 h)将CO2和环氧氯丙烷转化为对应的环状碳酸酯。通过延长反应时间(96 h),以酚羟基作为间隔单元的IP3可在较低温度(40℃)和pCO2=101 kPa条件下将环氧氯丙烷几乎定量地转化成环状碳酸酯,并且循环使用10次后,仍保持92%以上的产率,表现出优异的催化活性和循环稳定性,具有良好的应用前景。
1 实验部分
1.1 试剂和仪器
实验所用的试剂均为分析纯,所用水均为去离子水。1,3-二(溴甲基)苯、2,6-二甲基吡啶、N-(三甲基硅基)咪唑、环氧氯丙烷及其他环氧化合物、氯化亚砜、2,6-二(羟甲基)-4-甲基苯酚、偶氮二异丁腈(AIBN)和N-溴代琥珀酰亚胺(NBS)购自上海阿拉丁生化科技股份有限公司。所有药品和试剂不经任何处理直接使用。
实验中使用的仪器如下:红外光谱仪为Nicolet iS10(Thermo Nicolet),扫描范围是400~4 000 cm-1,分辨率为0.5 cm-1;扫描电子显微镜(SEM)型号是QUANTA FEG 450(美国FEI),工作电压为20 kV,通过喷金抽真空进行测试;X射线光电子能谱(XPS)用Escalab 250Xi(Thermo Fisher Scientific)X射线光电子能谱仪测试,激发光源为AlKα,测试区域为500 μm,测试压力低于10-7Pa;比表面积与孔隙率分析仪型号为MicroActive for ASAP 2460 2.01(美国麦克公司),样品真空预处理温度为100℃,氮气吸附测试时的吸附温度为77 K;能量色散X射线谱面扫(EDS Mapping)在X-Max能谱仪(英国牛津公司)上进行;气相色谱仪型号为7820A(美国安捷伦公司),载气(N2)压力为0.12 MPa,氢气压力为0.05 MPa,空气压力为0.05 MPa;NMR仪器型号为Varian 400-MR(美国安捷伦公司),以CDCl3作溶剂,四甲基硅作为内标。
1.2 2,6-二溴甲基吡啶(L2)的制备[18]
将 2,6-二甲基吡啶(0.536 g,5 mmol)置于 500 mL的烧瓶中,加入180 mL二氯甲烷/水混合溶剂(1:1,V/V)。然后在搅拌条件下加入NBS(3.0 g,16.8 mmol)和 AIBN(0.03 g,0.18 mmol),并置于油浴中加热,在红外灯(150 W)照射下回流24 h。反应完全后待反应液冷却至室温,进行萃取,所得下层有机相用无水Na2SO4干燥后进行减压浓缩。然后用乙酸乙酯/石油醚(1∶10,V/V)洗脱液进行柱色谱纯化,得到 淡 黄 色 固 体 (0.201 1 g,15.2%)。1H NMR(400 MHz):δ7.70(t,J=7.7 Hz,1H,Py),7.37(d,J=7.7 Hz,2H,Py),4.53(s,4H,2CH2)。13C NMR(101 MHz):δ156.7(Py),138.2(Py),122.9(Py),33.5(CH2)。
1.3 2,6-双(氯甲基)-4-甲酚(L3)的制备[19]
在冰浴条件下,将 SOCl2(8.7 mL,120 mmol)的THF(10 mL)溶液通过恒压滴液漏斗缓慢滴加到2,6-双(羟甲基)-4-甲基苯酚(3.36 g,20 mmol)的THF(30 mL)溶液中。待反应升到室温后,继续搅拌5 h。将反应液真空浓缩至除去所有溶剂,用水洗涤并用乙酸乙酯萃取(4×30 mL)。收集的有机溶液用无水Na2SO4干燥,真空浓缩后,采用二氯甲烷/石油醚混合溶剂冷冻重结晶的方法得到白色固体(0.773 g,85.9%)。1H NMR(400 MHz):δ7.09(s,2H,Ph),5.56(s,1H,OH),4.66(s,4H,2CH2),2.28(s,3H,CH3)。13C NMR(101 MHz):δ151.1(Ph),131.7(Ph),130.7(Ph),124.7(Ph),42.6(CH2),20.5(CH3)。
1.4 催化剂IP1~IP3的制备[20]
将 1,3-二(溴甲基)苯(L1,1.31 g,5 mmol)和N-(三甲基硅基)咪唑(0.70 g,5 mmol)溶解在MeCN中(20 mL)。将温度升至85℃,反应在氩气保护下回流48 h。反应完全后过滤除去溶剂和副产物,所得白色固体用MeCN(3×30 mL)和乙醚洗涤(5×50 mL)。在真空下干燥得到白色固体IP1(1.03 g,82.1%)。
其他催化剂制备过程与IP1类似,IP2和IP3制备所用的多卤化物分别是L2和L3。按此方法分别合成了离子聚合物IP2(0.798 g,63.3%)和IP3(1.02 g,86.4%)。
1.5 催化CO2转化合成环状碳酸酯反应
将一定质量的IP1/IP2/IP3和环氧氯丙烷(462.6 mg,5 mmol)加入至10 mL带支口的反应管中,无需加任何溶剂。反应管配有CO2气球并通过3次真空-CO2循环将气氛替换为CO2,反应在一定温度下进行,在一定的反应时间后,将反应管冷却至室温,取样用MeCN稀释后通过气相色谱测定产物产率。
1.6 催化剂的循环利用
根据催化活性比较,选择催化性能最优的催化剂IP3进行了催化剂循环使用测试。具体操作如下:将催化循环过的样品进行离心回收,经过4次无水乙醚洗涤,再离心并真空干燥后,进行下一次同样的催化实验,测试其可回收性及稳定性。
2 结果与讨论
2.1 离子聚合物IP1~IP3的合成
2,6-二溴甲基吡啶(L2)[18]和 2,6-双(氯甲基)-4-甲酚(L3)[19]通过文献报道的方法合成得到。聚合反应采用一锅直接季铵化法[20],以芳烃多卤化物和N-(三甲基硅基)咪唑作为前体,通过亲核取代反应形成重复的C—N键,可高产率地制得离子聚合物IP1~IP3(图1)。IP1和IP3为白色粉末状固体,IP2呈淡粉色。

图1 IP1~IP3的合成路线Fig.1 Synthetic routes of IP1-IP3
2.2 离子聚合物IP1~IP3的表征
2.2.1 红外光谱
制备的离子聚合物IP1~IP3的红外光谱如图2所示。从图中可以看出,IP1~IP3在3 141 cm-1处都出现了C—H伸缩振动,在2 970 cm-1处的吸收峰归因于亚甲基的C—H伸缩振动,证明骨架中咪唑环和吡啶环成功连接[21]。此外,1 155、1 561、1 625 cm-1处出现的吸收峰,分别为咪唑环上的C=N+伸缩振动、不饱和C=C伸缩振动以及C=N的伸缩振动,这证明骨架中咪唑阳离子的存在[20-21],也进一步证明了离子聚合物的成功合成。

图2 IP1~IP3的FT-IR谱图Fig.2 FT-IR spectra of IP1-IP3
2.2.2 SEM
图3是离子聚合物IP1~IP3的SEM图。图中显示出IP1~IP3都是由具有不规则形状和大小的团聚颗粒组成。由图3a和3b可以看出,离子聚合物IP1通过团聚物颗粒之间的紧密作用,相互交联形成大的聚合体,并且在交联过程中形成了一定的孔隙结构。从图3c和3d显示了IP2为不规则团聚颗粒多孔聚合物,表面粗糙且具有大量的孔。从图3e和3f可看出,离子聚合物IP3团聚颗粒之间的交联状态比IP1和IP2更紧密,这种独特的结构可能为CO2提供了丰富的吸收和反应位点。

图3 IP1(a、b)、IP2(c、d)和IP3(e、f)的SEM图Fig.3 SEM images of IP1(a,b),IP2(c,d),and IP3(e,f)
2.2.3 离子聚合物IP3的EDS Mapping、XPS和比表面积
我们选择了具有最优催化效果(见后文催化部分)的离子聚合物IP3进行了元素组成及分布表征。图4是IP3中C、N、O和Cl元素的EDS Mapping图,从图中可以看出这几种元素均匀分布在聚合物材料结构中。

图4 IP3中元素(C、N、O、Cl)的EDS Mapping图Fig.4 EDS Mapping images of the elements(C,N,O,and Cl)of IP3
通过XPS进一步对离子聚合物IP3的元素组成进行了测试(图5)。从图5b可以看出,N1s能谱图拟合曲线表明该样品中的N有2种不同的状态,其中401.3 eV的谱峰对应于咪唑的其中一个N与二卤化物相连所形成的N+,400.8 eV处的峰对应于咪唑的C—N键[21-22];而图5c中O1s谱图在532.4 eV处的特征峰来源于多卤化物L3的酚羟基,证明了这类离子聚合物的成功合成[23-24]。图5d中Cl2p的拟合曲线显示出IP3中的Cl存在2种不同的状态,197.2和196.8 eV分别对应于Cl-的Cl2p1/2和Cl2p3/2特征峰,Cl-含量较高;而198.7和198.0 eV则归属于C—Cl的Cl2p1/2和Cl2p3/2特征峰[22],其含量较低。这进一步表明大部分多卤化物L3发生了聚合。
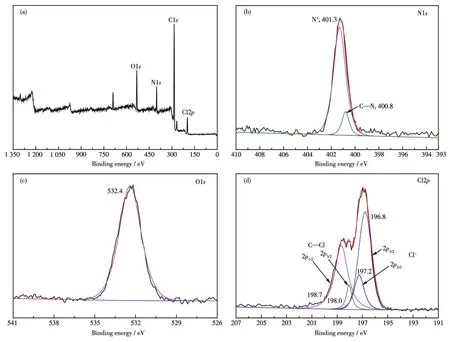
图5 IP3的XPS谱图Fig.5 XPS spectra of IP3
图6所示的N2吸附-脱附等温线显示离子聚合物IP3在较低的相对压力(p/p0<0.03)下N2吸附过程较为缓慢,而在较高的相对压力(0.03<p/p0<0.9)下,IP3对N2的吸附持续增加;此外,在相对压力(0.6<p/p0<1)下观察到明显的H3回滞环。该结果表明离子聚合物IP3对N2的吸附过程表现为明显的Ⅳ型等温线,并具有H3迟滞回线的特征,反映出该离子聚合物具有介孔结构。另一方面,IP3在对N2的解吸过程中表现出水分脱附的现象,这可能归因于该离子聚合物容易吸水,在完成对N2的脱附后,又进一步将聚合物骨架中的水分脱附出来。从IP3的孔径分布曲线图(图6插图)可以看出,IP3的孔径分布范围集中在3~50 nm之间,进一步验证了该离子聚合物材料中存在介孔结构。

图6 IP3的N2吸附-脱附等温线Fig.6 N2adsorption-desorption isotherm of IP3
2.3 离子聚合物IP1~IP3催化CO2与环氧化物的环加成反应
2.3.1 催化条件优化
为了探究离子聚合物IP1~IP3对CO2与环氧化合物环加成反应的催化性能,我们选择环氧氯丙烷作为反应底物(图7),分别对反应的最优催化剂、反应温度、催化剂用量以及反应溶剂进行了优化。

图7 以环氧氯丙烷为底物的CO2环加成反应Fig.7 Cycloaddition reaction of CO2 with epichlorohydrin as substrate
我们选取环氧氯丙烷转化成碳酸环氯丙烯酯作为模型反应,首先以二甲亚砜(DMSO)作为溶剂、在80℃和pCO2=101 kPa的反应条件下考察了离子聚合物IP1~IP3的催化性能。研究结果表明(表1,Entry 1~3),以酚羟基为间隔单元的IP3具有最好的催化活性,反应12 h后得到碳酸环氯丙烯酯的产率为72.6%,而以苯环、吡啶为间隔单元的IP1和IP2的催化产率分别为69.0%和54.0%。由于IP3中的间隔单元酚羟基是一种良好的氢键供体,在催化CO2的环加成反应中易于与环氧化合物形成氢键,从而活化环氧化合物,而以苯环、吡啶为间隔单元的IP1和IP2不能形成类似的氢键,这可能是IP3具有更好催化活性的原因[25-27]。

表1 不同催化剂、反应溶剂和反应温度对环加成反应的影响Table 1 Effect of catalyst,solvent and temperature on the cycloaddition reactiona
在确定离子聚合物IP3为最佳催化剂后,我们筛选了不同的反应溶剂,除了氯仿(CHCl3)和THF因为沸点较低选取了其对应的回流温度外,其他溶剂的反应温度均为80 ℃(表1,Entry 3~8)。结果显示强极性溶剂DMSO和N,N-二甲基甲酰胺(DMF)有利于该催化反应,产物产率分别为72.6%和54.0%,DMSO优于DMF,但在中等(MeCN和THF)和较弱极性(CHCl3和甲苯(PhMe))溶剂中催化反应几乎难以进行。这可能是由于在强极性溶剂DMSO和DMF中,IP3具有更好的分散性,有利于底物和催化剂的接触,促进催化反应的发生。有意思的是,该催化反应在无溶剂条件下表现出更为优异的催化性能,同等情况下IP3催化反应的产率达到96.8%(表1,Entry 9),而IP1和IP2在此条件的催化效果也得到极大改善,产率分别为93.5%和93.3%,产率分别增大了35.5%和72.8%。很显然,无溶剂条件成为该催化反应的最优选择。
接下来考察了反应温度的影响,100℃条件下的产率为97.3%(表1,Entry 12),与80 ℃差别不大。考虑到催化剂的实用性,我们进一步考察了这类离子聚合物在接近室温的40℃的催化性能。IP3在反应2 d的产率为58.3%,效果不理想,但通过延长反应时间到4 d,催化产率高达98.3%。在同样的条件下,IP1和IP2的催化产率也能达到97.7%和81.9%。该结果表明这类离子聚合物在温和条件下具有良好的催化性能。
在确定最优催化剂、反应温度、反应溶剂等反应条件后,我们又进一步探究了催化剂用量对反应的影响。在最优的反应条件下,我们分别测试了5、10、25、50和75 mg等不同量的催化剂IP3对反应的催化效果。从图8可以看出,5 mg催化剂IP3的产率仅为29.9%,增加催化剂用量至10 mg,产率急剧上升至68.2%,继续增加催化剂用量,产物产率稳步提升,而使用50 mg催化剂IP3得到产物的产率为98.6%,这意味着环氧氯丙烷几乎定量转化为碳酸环氯丙烯酯。尽管催化剂IP3用量增至75 mg时,碳酸环氯丙烯酯产率达到99.1%,但与50 mg用量时的产率相差不大,考虑节约成本的因素,最终确定催化剂用量50 mg作为最优用量。
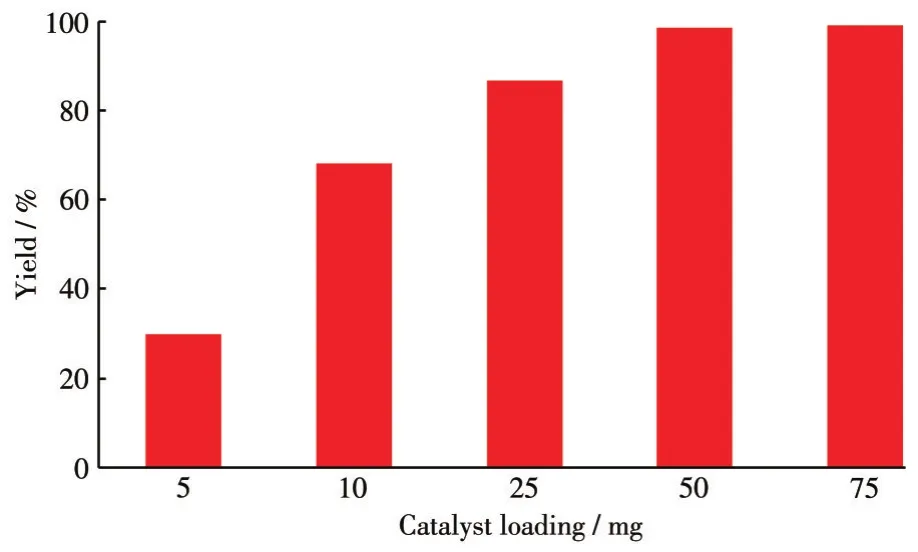
图8 催化剂IP3用量对环加成反应的影响Fig.8 Effect of catalyst IP3 loading on the cycloaddition reaction
2.3.2 底物拓展
在上述筛选的最优反应条件下,以离子聚合物IP3作为催化剂进行了多种环氧化物底物的拓展,实验结果列于表2。从中可以看出,IP3对含有卤素基团的环氧化物具有优异的催化活性(表2,Entry 1和2),在反应12 h后,几乎定量地将环氧氯丙烷和环氧溴丙烷转化为相应的环状碳酸酯。对于惰性的较长烷基链底物1,2-环氧辛烷,80℃反应条件下,该底物几乎没有任何的转化,但把温度升高至100℃时,却能定量地转化为对应的环状碳酸酯产物(表2,Entry 3)。类似的现象也出现在环氧环己烷的反应中(表2,Entry 4),但与 1,2-环氧辛烷不同的是,即使升温到100℃,产物的产率仅有11.0%,这可能归因于底物的位阻效应。位阻效应在底物苯基环氧乙烷的反应中也得以体现,该反应80℃时产率为41.2%,100℃时产率提升至98.2%(表2,Entry 5)。这些事实表明,在以IP3这类离子聚合物作为催化剂的CO2环加成反应中,底物的位阻效应对产物的形成起着重要的作用,类似的现象也发生在已报道的其他离子聚合物催化体系中[21,28-29]。对于含醚键的环氧化物底物,如烯丙基缩水甘油醚和环氧丙基苯基醚(表2,Entry 6和7),IP3也表现出良好的催化性能,可以中等以上收率(77.3%和68.7%)得到相应的环状碳酸酯。以上结果表明IP3在催化CO2环加成反应中具有良好的底物普适性。

表2 IP3催化CO2与不同环氧化物环加成反应的催化活性Table 2 Catalytic activity of IP3 for cycloaddition of CO2with different epoxidesa
2.3.3 循环稳定性
离子聚合物催化材料的一个优势是具有优异的催化循环稳定性。本研究中,我们选择了催化活性最好的离子聚合物IP3进行了催化剂的循环利用及稳定性测试实验,结果如图9所示。在上述最优的催化反应条件下,IP3循环使用10次后仍保持92%以上的产物产率,表明IP3具有优异的可循环性和稳定性。

图9 IP3的催化循环测试Fig.9 Recyclability tests of IP3
进一步地,我们通过SEM和FT-IR比较了离子聚合物IP3循环前后的变化。图10a和10b是IP3循环反应10次以后的SEM图,与图3e和3f新鲜制备的催化剂IP3相比,循环后的催化剂由之前的交联状态变为块状,这可能是由于IP3在循环反应过程中孔洞中吸附了一些小分子物质(溶剂、底物或产物),因此观察不到明显的孔隙结构。但孔的变化并没有影响催化剂的传质效率,IP3在相同的温度和反应时间条件下仍能得到与新鲜催化剂相当的优异产率。

图10 IP3循环反应10次后的SEM图Fig.10 SEM images of IP3 after 10 cycles of the reaction
从图11所示的红外光谱对照图可以看出,催化剂IP3在循环后仍在1 155 cm-1处具有明显的咪唑骨架C=N+特征峰,说明在循环过程中IP3的主体咪唑骨架没有被破坏,这也是催化性能在循环过程中得以维持的原因。相比较于新鲜制备IP3的FTIR谱图,循环后的IP3在1 793 cm-1处多了一个吸收峰,这归属于产物环状碳酸酯的C=O伸缩振动,原因在于在循环过程中,IP3的孔隙会吸附一些环状碳酸酯并且难以洗涤出来,这和上述SEM分析结果相一致。

图11 IP3循环反应10次前后的FT-IR谱图Fig.11 FT-IR spectra of IP3 before and after 10 cycles of the reaction
2.3.4 可能的催化机理
基于不同间隔单元离子聚合物IP1~IP3催化活性结果的对比,再结合已有文献报道的机理[25-27],我们提出了基于氢键供体活化环氧化合物促进CO2环加成的可能催化机理,如图12所示。该催化反应机理包含以下4个步骤:(1)环氧化合物与IP3中的酚羟基相互靠近形成氢键,形成环氧化合物被活化的中间体1。(2)IP3的Cl-亲核进攻环氧化合物位阻较小的碳原子,使得活化的环氧化合物开环形成中间体2。(3)CO2插入并与中间体2的氧负离子反应形成中间体3。(4)中间体3中的氧负离子进攻与氯相连的碳原子发生环合反应,生成产物环状碳酸酯,同时催化剂IP3得以再生。在整个催化循环过程中,离子聚合物IP3中含有的酚羟基可作为高效的氢键供体,既可以活化环氧化合物,又能稳定催化中间体2和3,同时IP3含有的氯离子可以作为亲核性较强且容易离去的亲核试剂。因此,IP3中含有的卤素离子和氢键供体在催化CO2环加成的过程中起到了“双活化”作用,大幅改善了催化效率,实现了较温和条件下的CO2高效转化。

图12 IP3催化CO2与环氧化合物环加成反应的可能机理Fig.12 Proposed mechanism for the reaction between epoxide and CO2catalyzed by IP3
3 结 论
通过一锅直接季铵化法构建了一类新型的离子聚合物IP1~IP3。由于IP1~IP3具有大量均匀分布的活性离子位点和丰富的介孔结构,将其应用于CO2与环氧化合物环加成反应时表现出优异的催化活性:在常压CO2气氛、无溶剂、80℃的温和条件下,3种离子聚合物均以超过93%的产率将环氧氯丙烷转化为碳酸环氯丙烯酯。相比较于苯环、吡啶作为间隔单元的IP1和IP2,酚羟基作为间隔单元的IP3无论在溶剂条件还是无溶剂条件下均表现出最佳的催化性能,表明间隔单元酚羟基在CO2环加成反应中可以作为重要的氢键供体,活化环氧化合物以促进反应的进行,从而提高催化效率。同时,离子聚合物IP3循环使用10次仍保持92%以上的产物收率,表明这类催化材料具有良好的循环稳定性。更为重要的是,在常压CO2气氛、40℃的温和条件下,通过适当延长反应时间,离子聚合物IP1~IP3仍能催化CO2环加成反应获得高产率,展现出这类聚合物催化材料的良好工业应用前景。
猜你喜欢
科学大众(2023年17期)2023-10-26 07:38:56
大电机技术(2022年5期)2022-11-17 08:14:04
云南化工(2021年6期)2021-12-21 07:30:56
小天使·二年级语数英综合(2021年5期)2021-07-11 10:58:35
科学(2020年2期)2020-08-24 07:57:00
世界农药(2019年4期)2019-12-30 06:25:14
中学化学(2017年2期)2017-04-01 08:51:54
材料科学与工程学报(2016年1期)2017-01-15 13:33:53
生物技术通报(2015年1期)2015-04-10 16:15:19
无机化学学报(2014年8期)2014-02-28 17:32:36

- 无机化学学报的其它文章
- La-Doped BaSnO3/Multi-walled Carbon Nanotube Modified Separator:Synthesis and Application in Lithium-Sulfur Battery
- Co(Ⅱ)/Ni(Ⅱ) Coordination Polymer of Isomeric Terphenyl-2,2″,4,4″-tetracarboxylic Acids with a Single Water Bridge:Syntheses,Structures,and Magnetic Properties
- Micromotors Based on Ni-Mn Binary Oxide and Its Application for Effective Dye Adsorption
- Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol by Mg-Doped Ceria Monolithic Catalyst
- Hydrogen Storage Capabilities of the Low-Lying Ca2B4Clusters
- 盘状镝簇合物的合成及缓慢磁弛豫