
能量代谢紊乱在高原心脏病发生过程中的作用*
2022-07-06 02:00韩艺玮张致英郝美莉张晓英
中国病理生理杂志 2022年6期
韩艺玮, 张致英, 郝美莉, 张晓英
能量代谢紊乱在高原心脏病发生过程中的作用*
韩艺玮, 张致英, 郝美莉, 张晓英△
(西藏民族大学医学院,西藏民族大学藏药检测技术教育部工程研究中心,陕西 咸阳 712082)
高原心脏病;动脉型肺动脉高压;右心室肥厚;能量代谢紊乱;缺氧诱导因子1α;过氧化物酶体增殖物激活受体
高原地区具有海拔高(≥3 000 m以上)、气压低、氧分压低的特点,易导致机体缺氧从而引发一系列急、慢性高原病,如急性肺水肿、高原红细胞增多症、缺氧性脑水肿、急慢性高原心脏病(high-altitude heart disease, HAHD)等,给高原地区居民人们的身心健康带来严重危害。其中慢性HAHD发病率高,危害大,不易逆转,预后差。更重要的是HAHD还会导致心脏、大脑和肾脏的衰竭,从而严重缩短寿命,是高海拔地区居民和平原移居人群必须重视的健康问题。
本文从动脉型肺动脉高压(pulmonary arterial hypertension, PAH)和右心室肥大(right ventricular hypertrophy, RVH)两个方面重点综述了能量代谢紊乱及其相关信号分子在HAHD发生、发展中的作用,旨在为HAHD发病机制的研究、疾病预防和治疗提供参考资料。
1 HAHD概述
HAHD是指正常人移居高原后,在长期低压、缺氧环境下,引起肺小动脉功能性和器质性改变,出现缺血性PAH和负荷性RVH,最后导致右心功能不全,至晚期则可出现左室肥厚扩大、全心衰以及心律失常等症状。据西藏医学研究所在4 500~4 700 m高原的调查显示,大约24.9%高原居民患有缺氧性RVH,明显RVH者如不能尽快干预最终会发展为HAHD[1]。
1.1PAHPAH是HAHD发生的主要因素,其血流动力学标准为:静息状态下,在海平面右心导管测量平均肺动脉压≥25 mmHg (1 mmHg=0.133 kPa)[2]。肺泡通气不足是PAH发生的始动因素。低压低氧的环境会导致肺泡过度通气,逐渐转变为相对低通气和肺泡通气不足,通气血流比值失调,导致肺泡和动脉血氧分压降低,进而造成缺氧通气反应[3]和缺氧性肺血管收缩[4]。研究显示,海拔越高,吸入气氧分压下降越显著,PAH的程度也越明显[5]。其次,慢性持续低氧可导致高原红细胞增多症,血液粘滞性增加,肺血容量增多,进一步加剧PAH的形成。
在PAH发生、发展中发挥决定性因素的是肺血管重构[6],即表现为:血管内皮损伤、中膜增厚、周围血管的肌纤维化和细胞外基质增多等,进而引起肺血管管腔狭窄、血管壁增厚等构象改变,甚至出现闭塞性病变[4, 6]。有报道指出,高原居民表现出RVH及肺血管压力增高,大多因为长期缺氧引起不可逆转的肺血管重构[7]。相似的结果也出现在动物模型中:高海拔低氧牛模型中肺血管表现出广泛的胶原沉积和血管中膜增厚重构[8];PAH小鼠模型出现明显的肺动脉肌化增加、血管闭塞和血管重塑等改变[9-10]。因此肺血管重构是PAH发生、发展的关键因素,导致RVH及右心功能进行性衰竭。
1.2RVH及右心功能不全RVH或右心功能不全作为HAHD的主要特征,同时也是PAH导致心脏后负荷增加的结果,具体表现为:右心室扩张,室间隔增厚,且随着右心房压力的不断增加,下腔静脉扩张,失去吸气性塌陷,会伴发一定程度的三尖瓣返流[9]。但也有研究显示,在HAHD发生的过程中,只出现RVH,并未存在扩张[10],导致不同的结果可能是由于采用的实验模型以及缺氧的程度和持续时间存在差异,特别是高海拔环境中的混杂因素所致。此外,需要重点关注的是PAH后负荷的长期存在,促使慢性RVH逐渐发展为心力衰竭[11]。其中氧化应激、炎症反应、心肌纤维化、凋亡、能量代谢紊乱等多种复杂的机制参与其中[12-13],而近年来能量代谢紊乱在HAHD发生过程中的作用逐渐受到重视。
2 能量代谢紊乱促进HAHD向心衰的发生
底物利用转变和线粒体功能障碍可促进肺动脉内皮细胞(pulmonary artery endothelial cells, PAECs)功能障碍和肺动脉平滑肌细胞(pulmonary artery smooth muscle cells, PASMCs)过度增殖,从而引发肺血管重构,导致PAH的发生,进而导致RVH及右心功能不全,而心肌细胞脂肪酸氧化能力的下降进一步加速了HAHD向心衰的进程。
2.1能量代谢紊乱促进PAH的发生发展肺血管重构作为PAH发生、发展的关键因素,与PAECs功能障碍、PASMCs过度增殖和凋亡失衡、炎症、血管过度收缩等密切相关[14]。其中PAECs功能障碍被认为是PAH病理生理学改变的关键起始因素[15],而PASMCs增殖与凋亡失衡是导致PAH的重要病因。
2.1.1底物利用转变促进PAECs增殖并加速PAH的发展PAECs糖酵解代谢途径的转变和过度增殖是PAH发生、发展的病理学基础。葡萄糖代谢在肺动脉高压PAECs的能量需求中发挥主要作用,当机体处于持续缺氧环境中时,葡萄糖的有氧代谢将逐渐转变为无氧糖酵解,PAECs中糖酵解速率较正常PAECs高出约3倍以上[16]。同时伴有丙酮酸脱氢酶激酶(pyruvate dehydrogenase kinases, PDK)的激活,PDK由4种同工酶(PDK1~PDK4)组成,可使线粒体葡萄糖氧化的关键酶丙酮酸脱氢酶(pyruvate dehydrogenase, PDH)磷酸化失活[17-18],促使新陈代谢从氧化磷酸化转向糖酵解[14]。
研究显示,PAECs中的葡萄糖转运蛋白1上调[19],6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶3(6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, PFKFB3)升高[20],乳酸积累,从而导致PAECs功能障碍,进而损伤肺组织的结构和功能[21]。过表达的亦可使患有PAH的啮齿动物和人群肺血管内皮中生长因子和促炎因子分泌增加,这将进一步促进PAECs的过度增殖。
2.1.2活性氧簇(reactive oxygen species, ROS)和PASMCs在PAH形成过程中的影响ROS是一种具有潜在毒性的超氧化物,线粒体复合物I和III是其产生的重要位点[22]。ROS可被超氧化物歧化酶2(superoxide dismutase-2, SOD-2;一种仅存在于线粒体中的酶)转化为可扩散的第二信使过氧化氢(hydrogen peroxide, H2O2)。而H2O2参与氧敏感的电压门控K+通道(voltage-gated potassium channels, Kv;例如Kv1.5和Kv2.1)的激活和表达[23-24]。
PASMCs是构成肺动脉壁的主要细胞[25]。在PAH患者和缺氧条件下培养PASMCs,线粒体细胞中ROS的释放和积累导致高海拔大鼠PASMCs中线粒体膜Kv通道的功能降低[14]和线粒体膜电位去极化,这不仅使细胞内K+浓度升高,抑制促凋亡的胱天蛋白酶,抑制细胞凋亡,而且导致电压门控L型钙通道激活,大量的钙内流促进细胞增殖[23-24, 26]。此外,ROS还可导致细胞色素C氧化酶和SOD水平降低,从而破坏线粒体代谢功能和酶动力学活性,加速PASMCs增殖并抑制细胞凋亡[27]。这种线粒体细胞内ROS的积累导致PASMCs的增殖与凋亡失衡可能与PAECs线粒体ROS产生激活存在一致性,协同促进了PAH的发生发展。
2.2能量代谢紊乱促进RVH及右心功能不全心脏作为高耗能、高耗氧器官,对能量的需求远高于一般组织器官,而心肌细胞能量代谢紊乱直接或间接促进了HAHD中RVH的发生。
2.2.1能量代谢重构促进HAHD心脏的肥大和衰竭在高原低压、低氧环境下,机体除了利用增加红细胞的数量来增加氧气输送外,还通过代谢调节来提高组织对氧气的利用率,如降低心脏磷酸肌酸与三磷酸腺苷(adenosine triphosphate, ATP)比率,抑制脂肪酸氧化和增加心脏葡萄糖摄取利用[28]。但是氧气利用率的提高并不能完全满足机体对氧气的需求,尤其在高原持续低氧暴露下,PDK1在右心室表达明显增加[29],进一步抑制丙酮酸进入三羧酸循环,从而使代谢向生成乳酸的糖酵解方向进行[30]。而这种长期以糖酵解为主要供能机制的代谢紊乱,使心肌细胞不得不为了维持泵血而进行结构和功能的慢性重塑,最终发展为HAHD。
2.2.2高原低氧通过抑制过氧化物酶体增殖物激活受体α(peroxisome proliferator-activated receptor α, PPARα)、激活PPARγ促进心肌细胞代谢转变正常情况下,脂肪酸氧化产生的ATP占心脏ATP含量的70%[31]。PPARα是调节细胞能量代谢、脂质代谢和维持心肌能量代谢稳态的关键因子,其转录调控与高原缺氧的心脏功能存在联系[32]。缺氧环境会明显抑制心肌中PPARα的表达[33],导致心肌细胞中脂肪酸摄取和β氧化减少。Murray等[28]和Horscroft等[34]在对喜马拉雅夏尔巴人和藏族高海拔居住人群的研究中观察到了类似的结果,下调表达可以降低脂肪酸氧化能力,进而增加氧气的利用率。但是高原缺氧环境下PPARα持续减少,心脏长期处于应对低氧的高负荷状态,脂肪酸到糖酵解的代谢转换可能不足以维持心肌正常的能量代谢和高能磷酸盐(Ca2+)含量,导致心脏无法维持高收缩性能和射血功能,进而造成心肌细胞的超负荷工作,引起高原性心脏肥大和衰竭[35],更有甚者心肌出现病理性纤维化的表型[36],形成不可逆的HAHD。
研究表明,PPARγ虽是脂肪生成所必需的,但也存在于心血管系统中[37],是高海拔适应的候选因子[38]。Krishnan等[39]和Yu等[40]观察到缺氧以及肥厚型心肌病的人和小鼠的心室活检样本中缺氧诱导因子1α(hypoxia-inducible factor 1α, HIF-1α)和PPARγ的表达都是增加的。
总之,高原心脏中PPARα减少引起的脂肪酸氧化能力降低伴随着PPARγ的激活,在血流动力学超负荷期间,心脏功能更容易受到损伤和器质性重构[28, 35],逐渐向肥大和衰竭发展。
3 能量代谢信号通路促进HAHD
能量代谢改变是HAHD发生的重要原因之一,而HIF-1α、PPARα和PDK与HAHD的能量代谢调节存在联系,因此对相关能量代谢信号通路在HAHD发生发展过程中潜在机制的阐述具有重要意义。
3.1HIF-1α促进PAH的发生发展HIF-1α作为应对缺氧的关键调节因子,其激活会增加HAHD中PAECs和PASMCs增殖和肺血管重塑,促进PAH的发生发展。
3.1.1HIF-1α参与HAHD中PAECs的糖酵解慢性缺氧刺激下,HIF-1α作为应对缺氧的关键调节因子被激活[41],其激活会增加许多糖酵解酶以及PDK的表达,促使氧化代谢不断向糖酵解转换[42],导致HAHD中PAECs的过度增殖。具体机制为:HIF-1α不仅通过激活PDK表达,损害氧化代谢,促进糖酵解代谢,而且还可诱导PFKFB3的mRNA和蛋白表达,促进PAECs的糖酵解和过度增殖[43]。
此外,PAECs中白细胞介素33(interleukin-33, IL-33)及其受体ST2的免疫反应性的显著增加可能会导致PAH。缺氧会诱导PAECs的IL-33/ST2表达升高,激活HIF1α/血管内皮生长因子(vascular endothelial growth factor, VEGF)轴,诱导PAECs的血管生成和增殖,从而导致PAH[44],并可能为PASMCs重塑的启动提供基础[45]。这表明炎症进一步促进了PAECs的过度增殖。
3.1.2HIF-1α促进HAHD中PASMCs的增殖和肺血管重塑在HAHD发病过程中,缺氧导致的HIF-1α激活决定了PASMCs的增殖和肺血管重塑[46]。其中缺氧环境下ROS的积累和释放以及第二信使H2O2的减少,会导致HIF-1α激活,抑制氧敏感Kv通道的表达,促进PASMCs的增殖与凋亡失衡,导致PAH的发生[14]。线粒体在缺氧性PAH的产生中仍起着关键作用。其具体机制为:NADH脱氢酶(泛醌)1α亚复合物4样2[NADH dehydrogenase (ubiquinone) 1α subcomplex 4 like 2, NDUFA4L2]作为电子传递链复合物I亚基的一部分,通过调节上游HIF-1α和下游p38-5-脂氧合酶(5-lipoxygenase, 5-LO)信号促进PASMCs的增殖,从而促进肺血管重塑,诱导PAH[47]。
另有研究报道,黏附受体CD146和HIF-1α交叉调节是血管重塑和PAH发病的关键因素,PASMCs中CD146-HIF-1α轴的破坏会削弱肺血管重塑,这揭示了血管重塑过程中的缺氧重编程[48]。
3.2PPARα和PDK调节HAHD心脏的能量代谢PPARα和PDK作为调节HAHD心脏能量代谢的关键受体和激酶,二者的异常均可引发能量代谢紊乱,从而加速HAHD的发生发展。
3.2.1PPARα介导HAHD心脏能量代谢转换Narravula等[49]报道,缺氧心肌细胞的调控与PPARα和PPARγ有关,PPARα的下调可能是心肌细胞缺氧期间的适应性反应。反之,PPARγ的激活促进了缺氧大鼠心脏心房钠尿肽的分泌[40, 50],并将能量代谢的途径向糖酵解的方向进行重编程[51],而其中PPARα可能是HAHD发生发展的核心受体。
研究表明,微小RNA(microRNA, miRNA, miR)、Krüppel样因子5(Krüppel-like factor 5, KLF5)等可通过抑制PPARα发挥调控心脏能量代谢的作用[52]。miR-148a与miR-17-5p协同抑制PPARα,降低脂肪酸代谢[53]。miR-21也能有效降低的表达,抑制脂肪酸氧化[54],导致代谢转换至糖酵解,促进心肌细胞结构和功能的慢性重塑,最终发展为HAHD。此外,Drosatos等[31]报道,KLF5是心脏的正转录调节因子,其抑制导致心脏脂肪酸氧化减少和甘油三酯积累增加,从而导致心脏功能障碍。
SIRT3是一种定位于线粒体的NAD+依赖性蛋白质赖氨酸脱乙酰酶,在高海拔地区因线粒体功能变化而降低,进而降低三羧酸循环和ATP的生成[55-56]。SIRT3和PPARα都是HAHD线粒体稳态的关键调节因子。体内和体外研究表明,PPARα是SIRT3的上游转录调控因子[57]。Zong等[58]通过染色质免疫沉淀和萤光素酶实验也进一步证明了SIRT3是PPARα的直接下游靶点。总之,PPARα可能通过SIRT3进一步促进了HAHD心脏的能量代谢转换。
3.2.2HIF-1α通过PDK介导HAHD心衰的发生发展心衰是HAHD发展的最终结果。右心衰患者HIF-1α直接激活编码PDK1的基因,促使PDH失活,抑制三羧酸循环介导心肌代谢向糖酵解转变[59],从而损害右心室功能[60]。但并非所有的结论存在一致性,Piao等[60]检测到心脏亚型PDK2和PDK4的上调,而PDK1和PDK3不变,认为转录因子FOXO1介导了PDK4激活,而抑制PDK可改善右心室功能和运动能力。最后,我们总结和梳理了HAHD中与能量代谢紊乱相关的信号通路,详见图1。
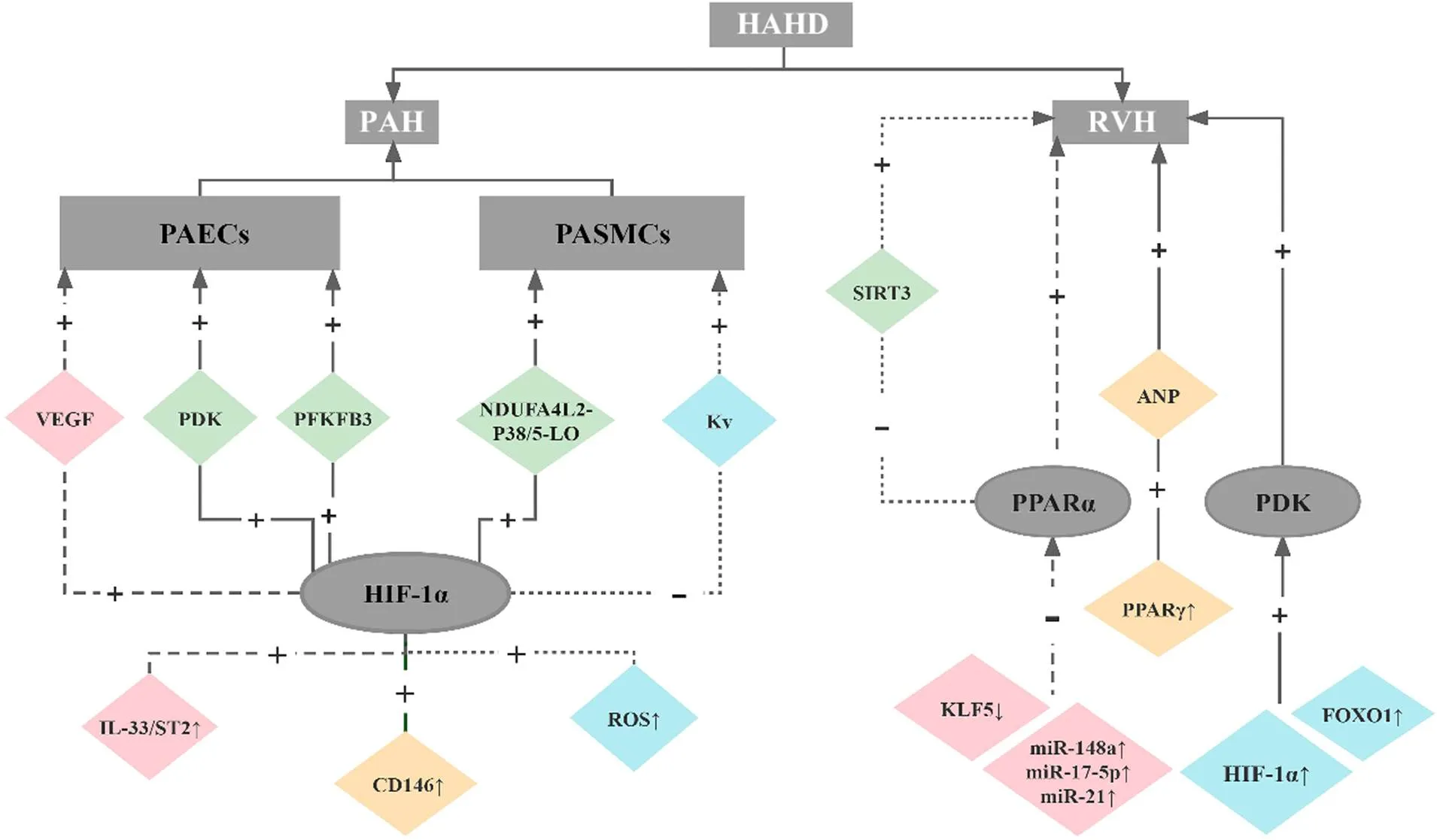
Figure 1. The signaling pathways of energy metabolism disorder in high-altitude heart disease (HAHD). HAHD is characterized by pulmonary arterial hypertension (PAH) and right ventricular hypertrophy (RVH). As the principal cause of PAH, excessive proliferation of pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs) based on hypoxia-inducible factor 1α (HIF-1α) is induced by activation of several signaling pathways, such as interleukin-33 (IL-33)/ST2 (IL-33 receptor)-HIF-1α-vascular endothelial growth factor (VEGF) and reactive oxygen species (ROS)-HIF-1α-voltage-gated potassium channels (Kv). Morover, peroxisome proliferator-activated receptor α (PPARα) inhibition and pyruvate dehydrogenase kinase (PDK) activation promote the development of RVH.
4 HAHD的治疗
基于上述对HAHD发生发展的机制探讨,相关激动剂和抑制剂的研究开始趋向于通过调节能量代谢来改善HAHD。
4.1PDK抑制剂二氯乙酸盐(dichloroacetate, DCA)作为PDK的抑制剂,可激活线粒体酶PDH(葡萄糖氧化的守门酶),逆转慢性缺氧性PAH,提高线粒体功能。这种表观遗传的线粒体代谢途径是一个潜在的抗纤维化治疗靶点[61]。线粒体异常中的Kv通道功能障碍的逆转也可以通过PDK抑制剂DCA与肺血管重塑的消退相关联来实现[14]。重要的是,DCA可抑制FOXO1诱导的PDK4上调并恢复心输出量,从而减轻PVH[60]。
4.2PPARα/γ激动剂PPARα和PPARγ激动剂已被宣称可用于治疗代谢性疾病和心血管疾病[62-63],其中PPARα是治疗血脂异常和相关心血管并发症的理想靶点[64]。神奇的是PPARγ激动剂吡格列酮的口服治疗完全逆转了实验性大鼠的PAH和血管重塑,有效改善了心室的结构和功能,抑制了右心衰[65]。但是它们对心肌的直接作用也有可能导致心脏功能恶化[66]。所以研发比现有PPARα/γ激动剂更高激动活性和选择性的激动剂,可作为治疗HAHD患者的潜在药物。
5 总结和展望
综上所述,HIF-1α通过增加PDK和PFKFB3的表达促进PAECs的糖酵解和过度增殖;通过抑制Kv通道和增加NDUFA4L2的表达导致PASMCs的增殖与凋亡失衡,进一步促进PAH的发生发展。PPARα作为HAHD发生发展的核心受体,可通过miRNA、KLF5和SIRT3等调节因子调控心脏能量代谢向糖酵解的方向进行重编程,而由HIF-1α和FOXO1激活的PDK更是加速了HAHD心衰的发生。上述研究表明阻断能量代谢紊乱中的相关通路可作为开发治疗HAHD药物的重要基石,其中具有高选择性和敏感性的新型PDK抑制剂和PPARs激动剂有可能成为具有前景的治疗HAHD的新药物。
[1] Xie ZZ, Liu FY, Wang SJ, et al. Preventive and therapeutic effects of nitrendipine on hypoxic right ventricular hypertrophy[J]. Acta Pharmacol Sin, 1996, 17(4):337-340.
[2] Soria R, Egger M, Scherrer U, et al. Pulmonary arterial pressure at rest and during exercise in chronic mountain sickness: a meta-analysis[J]. Eur Respir J, 2019, 53(6):1802040.
[3] Wagner PD, Wagner HE, Groves BM, et al. Hemoglobin P50during a simulated ascent of Mt. Everest, Operation Everest II[J]. High Alt Med Biol, 2007, 8(1):32-42.
[4] Grünig E, Mereles D, Hildebrandt W, et al. Stress Doppler echocardiography for identification of susceptibility to high altitude pulmonary edema[J]. J Am Coll Cardiol, 2000, 35(4):980-987.
[5] Ma TT, Wang Y, Zhou XL, et al. Research on rat models of hypobaric hypoxia-induced pulmonary hypertension[J]. Eur Rev Med Pharmacol Sci, 2015, 19(19):3723-3730.
[6] Oswald-Mammosser M, Weitzenblum E, Quoix E, et al. Prognostic factors in COPD patients receiving long-term oxygen therapy. Importance of pulmonary artery pressure[J]. Chest, 1995, 107(5):1193-1198.
[7] Hakim TS, Michel RP, Minami H, et al. Site of pulmonary hypoxic vasoconstriction studied with arterial and venous occlusion[J]. J Appl Physiol Respir Environ Exerc Physiol, 1983, 54(5):1298-1302.
[8] Penaloza D, Arias-Stella J. The heart and pulmonary circulation at high altitudes: healthy highlanders and chronic mountain sickness[J]. Circulation, 2007, 115(9):1132-1146.
[9] Naeije R. Physiological adaptation of the cardiovascular system to high altitude[J]. Prog Cardiovasc Dis, 2010, 52(6):456-466.
[10] Pratali L, Allemann Y, Rimoldi SF, et al. RV contractility and exercise-induced pulmonary hypertension in chronic mountain sickness: a stress echocardiographic and tissue Doppler imaging study[J]. JACC Cardiovasc Imaging, 2013, 6(12):1287-1297.
[11] Li X, Zhang Q, Nasser MI, et al. Oxygen homeostasis and cardiovascular disease: a role for HIF?[J]. Biomed Pharmacother, 2020, 128:110338.
[12] Dewachter L, Dewachter C. Inflammation in right ventricular failure: does it matter?[J]. Front Physiol, 2018, 9:1056.
[13] Tham YK, Bernardo BC, Ooi JY, et al. Pathophysiology of cardiac hypertrophy and heart failure: signaling pathways and novel therapeutic targets[J]. Arch Toxicol, 2015, 89(9):1401-1438.
[14] Bonnet S, Michelakis ED, Porter CJ, et al. An abnormal mitochondrial-hypoxia inducible factor-1α-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension[J]. Circulation, 2006, 113(22):2630-2641.
[15] Eddahibi S, Morrell N, d'Ortho MP, et al. Pathobiology of pulmonary arterial hypertension[J]. Eur Respir J, 2002, 20(6):1559-1572.
[16] Xu W, Koeck T, Lara AR, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells[J]. Proc Natl Acad Sci U S A, 2007, 104(4):1342-1347.
[17] Plecitá-Hlavatá L, Tauber J, Li M, et al. Constitutive reprogramming of fibroblast mitochondrial metabolism in pulmonary hypertension[J]. Am J Respir Cell Mol Biol, 2016, 55(1):47-57.
[18] Michelakis ED, Gurtu V, Webster L, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients[J]. Sci Transl Med, 2017, 9(413):eaao4583.
[19] Marsboom G, Wietholt C, Haney CR, et al. Lung ¹⁸F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension[J]. Am J Respir Crit Care Med, 2012, 185(6):670-679.
[20] Cao Y, Zhang X, Wang L, et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension[J]. Proc Natl Acad Sci U S A, 2019, 116(27):13394-13403.
[21] Mulukutla BC, Yongky A, Le T, et al. Regulation of glucose metabolism-aperspective from cell bioprocessing[J]. Trends Biotechnol, 2016, 34(8):638-651.
[22] Michelakis ED, Hampl V, Nsair A, et al. Diversity in mitochondrial function explains differences in vascular oxygen sensing[J]. Circ Res, 2002, 90(12):1307-1315.
[23] Archer SL, Wu XC, Thébaud B, et al. Preferential expression and function of voltage-gated, O2-sensitive K+channels in resistance pulmonary arteries explains regional heterogeneity in hypoxic pulmonary vasoconstriction: ionic diversity in smooth muscle cells[J]. Circ Res, 2004, 95(3):308-318.
[24] Archer SL, Souil E, Dinh-Xuan AT, et al. Molecular identification of the role of voltage-gated K+channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes[J]. J Clin Invest, 1998, 101(11):2319-2330.
[25] Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension[J]. J Clin Invest, 2012, 122(12):4306-4313.
[26] Prathapan A, Varghese MV, Abhilash S, et al. Polyphenol rich ethanolic extract fromL. mitigates angiotensin II induced cardiac hypertrophy and fibrosis in rats[J]. Biomed Pharmacother, 2017, 87:427-436.
[27] Wu D, Dasgupta A, Read AD, et al. Oxygen sensing, mitochondrial biology and experimental therapeutics for pulmonary hypertension and cancer[J]. Free Radic Biol Med, 2021, 170:150-178.
[28] Murray AJ,Montgomery HE, Feelisch M, et al. Metabolic adjustment to high-altitude hypoxia: from genetic signals to physiological implications[J]. Biochem Soc Trans, 2018, 46(3):599-607.
[29] Adrogue JV, Sharma S, Ngumbela K, et al. Acclimatization to chronic hypobaric hypoxia is associated with a differential transcriptional profile between the right and left ventricle[J]. Mol Cell Biochem, 2005, 278(1/2):71-78.
[30] Murray AJ. Energy metabolism and the high-altitude environment[J]. Exp Physiol, 2016, 101(1):23-27.
[31] Drosatos K, Pollak NM, Pol CJ, et al. Cardiac myocyte KLF5 regulatesexpression and cardiac function[J]. Circ Res, 2016, 118(2):241-253.
[32] Yang J, Liu C, Jihang Z, et al.genetic variants increase the risk for cardiac pumping function reductions following acute high-altitude exposure: a self-controlled study[J]. Mol Genet Genomic Med, 2019, 7(10):e00919.
[33] Yan J, Song K, Bai Z, et al. WY14643 improves left ventricular myocardial mitochondrial and systolic functions in obese rats under chronic persistent hypoxia via the PPARα pathway[J]. Life Sci, 2021, 266:118888.
[34] Horscroft JA, Kotwica AO, Laner V, et al. Metabolic basis to Sherpa altitude adaptation[J]. Proc Natl Acad Sci U S A, 2017, 114(24):6382-6387.
[35] Luptak I, Balschi JA,Xing Y, et al. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-α-null hearts can be rescued by increasing glucose transport and utilization[J]. Circulation, 2005, 112(15):2339-2346.
[36] Watanabe K, Fujii H, Takahashi T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator-activated receptor α associated with age-dependent cardiac toxicity[J]. J Biol Chem, 2000, 275(29):22293-22299.
[37] Duan SZ, Ivashchenko CY, Russell MW, et al. Cardiomyocyte-specific knockout and agonist of peroxisome proliferator-activated receptor-γ both induce cardiac hypertrophy in mice[J]. Circ Res, 2005, 97(4):372-379.
[38] Liu XL, Zhang FL, Zhou ZY, et al. Analysis of two sequence variants in peroxisome proliferator activated receptor γ gene in Tajik population at high altitudes and Han population at low altitudes in China[J]. Mol Biol Rep, 2010, 37(1):179-184.
[39] Krishnan J, Suter M, Windak R, et al. Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy[J]. Cell Metab, 2009, 9(6):512-524.
[40] Yu L, Li W, Park BM, et al. Hypoxia augments NaHS-induced ANP secretion via KATPchannel, HIF-1α and PPAR-γ pathway[J]. Peptides, 2019, 121:170123.
[41] Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress[J]. Mol Cell, 2010, 40(2):294-309.
[42] Cerychova R, Pavlinkova G. HIF-1, metabolism, and diabetes in the embryonic and adult heart[J]. Front Endocrinol (Lausanne), 2018, 9:460.
[43] Wei L, Zhang L, Yang L, et al. Protective effect of mesenchymal stem cells on isolated islets survival and against hypoxia associated with the HIF-1α/PFKFB3 pathway[J]. Cell Transplant, 2022, 31:9636897211073127.
[44] Liu J, Wang W, Wang L, et al. IL-33 initiates vascular remodelling in hypoxic pulmonary hypertension by up-regulating HIF-1α and VEGF expression in vascular endothelial cells[J]. EBioMedicine, 2018, 33:196-210.
[45] Kumar R, Graham B. IL-33-HIF1α axis inhypoxic pulmonary hypertension[J]. EBioMedicine, 2018, 33:8-9.
[46] Wilkins MR, Ghofrani HA, Weissmann N, et al. Pathophysiology and treatment of high-altitude pulmonary vascular disease[J]. Circulation, 2015, 131(6):582-590.
[47] Liu Y, Nie X, Zhu J, et al. NDUFA4L2 in smooth muscle promotes vascular remodeling in hypoxic pulmonary arterial hypertension[J]. J Cell Mol Med, 2021, 25(2):1221-1237.
[48] Luo Y, Teng X, Zhang L, et al. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension[J]. Nat Commun, 2019, 10(1):3551.
[49] Narravula S, Colgan S. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor α expression during hypoxia[J]. J Immunol, 2001, 166(12):7543-7548.
[50] Li X, Zhang Y, Zhang B, et al. HIF-1α-L-PGDS-PPARγ regulates hypoxia-induced ANP secretion in beating rat atria[J]. Prostaglandins Other Lipid Mediat, 2018, 134:38-46.
[51] Feng J, Dai W, Mao Y, et al. Simvastatin re-sensitizes hepatocellular carcinoma cells to sorafenib by inhibiting HIF-1α/PPAR-γ/PKM2-mediated glycolysis[J]. J Exp Clin Cancer Res, 2020, 39(1):24.
[52] Madrazo JA, Kelly DP. The PPAR trio: regulators of myocardial energy metabolism in health and disease[J]. J Mol Cell Cardiol, 2008, 44(6):968-975.
[53] Chen Z, Luo J, Sun S, et al. miR-148a and miR-17-5p synergistically regulate milk TAG synthesis viaandin goat mammary epithelial cells[J]. RNA Biol, 2017, 14(3):326-338.
[54] Chung KW, Lee EK, Lee MK, et al. Impairment of PPARα and the fatty acid oxidation pathway aggravates renal fibrosis duringaging[J]. J Am Soc Nephrol, 2018, 29(4):1223-1237.
[55] Murray AJ, Horscroft JA. Mitochondrial function at extreme high altitude[J]. J Physiol, 2016, 594(5):1137-1149.
[56] Dikalova AE, Pandey A, Xiao L, et al. Mitochondrial deacetylase Sirt3 reduces vascular dysfunction and hypertension while Sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress[J]. Circ Res, 2020, 126(4):439-452.
[57] Chen X, Wang Q, Shao M, et al. Ginsenoside Rb3 regulates energy metabolism and apoptosis in cardiomyocytes via activating PPARα pathway[J]. Biomed Pharmacother, 2019, 120:109487.
[58] Zong X, Cheng K, Yin G, et al. SIRT3 is a downstream target of PPAR-α implicated in high glucose-induced cardiomyocyte injury in AC16 cells[J]. Exp Ther Med, 2020, 20(2):1261-1268.
[59] Piao L, Fang YH, Cadete VJ, et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle[J]. J Mol Med (Berl), 2010, 88(1):47-60.
[60] Piao L, Sidhu VK, Fang YH, et al. FOXO1-mediated upregulation of pyruvate dehydrogenase kinase-4 (PDK4) decreases glucose oxidation and impairs right ventricular function in pulmonary hypertension: therapeutic benefits of dichloroacetate[J]. J Mol Med (Berl), 2013, 91(3):333-346.
[61] Tian L, Wu D, Dasgupta A, et al. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: a pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis[J]. Circ Res, 2020, 126(12):1723-1745.
[62] Han L, Shen WJ, Bittner S, et al. PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-β/δ and PPAR-γ[J]. Future Cardiol, 2017, 13(3):279-296.
[63] Takada I, Makishima M. Peroxisome proliferator-activated receptor agonists and antagonists: a patent review (2014-present)[J]. Expert Opin Ther Pat, 2020, 30(1):1-13.
[64] Han L, Shen WJ, Bittner S, et al. PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α[J]. Future Cardiol, 2017, 13(3):259-278.
[65] Legchenko E, Chouvarine P, Borchert P, et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation[J]. Sci Transl Med, 2018, 10(438):eaao0303.
[66] Son NH, Park TS, Yamashita H, et al. Cardiomyocyte expression of PPARγ leads to cardiac dysfunction in mice[J]. J Clin Invest, 2007, 117(10):2791-2801.
Role of energy metabolism disorder in development of high-altitude heart disease
HAN Yi-wei, ZHANG Zhi-ying, HAO Mei-li, ZHANG Xiao-ying△
(,,,,712082,)
High-altitude heart disease (HAHD) is a chronic progressive disease characterized by pulmonary arterial hypertension (PAH) and right ventricular hypertrophy (RVH) or right heart dysfunction caused by low pressure and hypoxia. As the main pathological feature of HAHD, RVH can be caused by oxidative stress, inflammation, fibrosis, energy metabolism disorder and other mechanisms. Energy metabolism disorder, as one of the pathogenic factors of PAH and RVH, has attracted much attention in recent years. As transcriptional regulatory factors affecting energy metabolism, hypoxia-inducible factor 1α (HIF-1α) and peroxisome proliferator-activated receptor α (PPARα) are involved in the regulation of PDK, PFKFB3, Kv, NDUFA4L2, miRNAs, KLF5, SIRT3, FOXO1 and other factors to promote the development of HAHD. In this paper, we review the research progress of the role of HIF-1α and PPARα/γ in HAHD.
High-altitude heart disease; Pulmonary arterial hypertension; Right ventricular hypertrophy; Energy metabolism disorder; Hypoxia-inducible factor 1α; Peroxisome proliferator-activated receptors
R541.9; R363.2
A
10.3969/j.issn.1000-4718.2022.06.022
1000-4718(2022)06-1135-07
2021-10-29
2022-05-07
西藏自治区自然科学基金项目[No. XZ2018ZR G-85(Z)];陕西省教育厅专项科学研究计划(No. 19JK0890);西藏民族大学重大项目培育计划(No. 18MDZ03; No. 20MDT03);西藏自治区自然科学基金资助项目(No. XZ202001ZR0089G)
Tel: 029-33755247; E-mail: melani1983@126.com
(责任编辑:宋延君,罗森)
猜你喜欢
华夏医学(2021年1期)2021-12-05
中国慈善家(2021年5期)2021-11-19
心肺血管病杂志(2020年5期)2021-01-14
英语文摘(2019年6期)2019-09-18
体育科学(2018年12期)2019-01-04
商周刊(2018年11期)2018-06-13
读与写·上旬刊(2018年1期)2018-05-06
教育教学论坛(2016年51期)2017-03-22
教育教学论坛(2016年49期)2017-02-27
科学中国人(2016年9期)2016-11-04
