
基于异质图注意力网络的miRNA与疾病关联预测算法
2022-07-02 06:21李政伟李佳树尤著宏钟堂波
电子学报 2022年6期
李政伟,李佳树,尤著宏,聂 茹,赵 欢,钟堂波
(1. 中国矿业大学矿山数字化教育部工程研究中心,江苏徐州 221116;2. 中国矿业大学计算机科学与技术学院,江苏徐州221116;3. 西北工业大学计算机学院,陕西西安 710129)
1 引言
MicroRNA(缩写为miRNA)是一类小的、内源性、非编码单链RNA 分子,其长度大约为22 个核苷酸,在人类蛋白质编码基因的调控中起到关键作用[1]. 诸多研究分析显示miRNA 在众多生物进程中,例如细胞增殖、分化、凋亡、病毒感染等[2],起着至关重要的作用.同时,miRNA 的突变或者异常表达往往会诱导多种人类复杂疾病的产生和演化[3]. 例如,通过单变量Cox 回归分析发现,miR-155 和miR-150 的表达水平对淋巴瘤病人的无进展生存期(Progression-Free-Survival,PFS)有着重要影响[4]. 因此,识别miRNA 与疾病间的潜在关联有助于医疗人员从分子角度理解疾病的病理机理,从而促进临床诊断、治疗和预后.
传统的识别miRNA 与疾病间潜在关联的生物学湿实验方法主要有Northern 杂交[5]、逆转录聚合酶链反应[6]、微阵列分析[7]等. 但是这些方法往往会受到环境影响,且需要大量的资金和时间投入,效率低下. 随着计算机的存储和运算能力的飞速发展,以及大量收集相关miRNA 和疾病信息的生物数据库的建立,设计更加高效的计算方法,实现大规模、高置信度地预测miRNA 与疾病间的潜在关联,逐渐受到科研人员的广泛关注[8,9].
启发于深度学习理论在生物信息学领域的成功应用[10,11],本文提出一种基于异质图注意力网络的端到端模型即HGATMDA(Heterogeneous Graph Attention Network for MiRNA-Disease Associations Prediction)来预测miRNA 与疾病间的潜在关联. 具体而言,首先将集成的miRNA 相似性信息、集成的疾病相似性信息以及经实验验证的miRNA-疾病关联整合进miRNA-疾病异质图中,并设计了顶点类型转换矩阵将异质的顶点特征投影至同一向量空间中;其次,采用多头注意力机制聚合异质邻居顶点特征,并将聚合后的特征与中心顶点的属性特征相融合,得到更具有表达能力的miRNA和疾病顶点的特征表示;之后,将miRNA-疾病对特征输入至全连接层(Fully Connected Layer,FCL)中得出预测的概率;最后,根据预测的概率与标签间的损失对整个模型进行端到端的训练. HGATMDA 模型的流程图如图1所示.
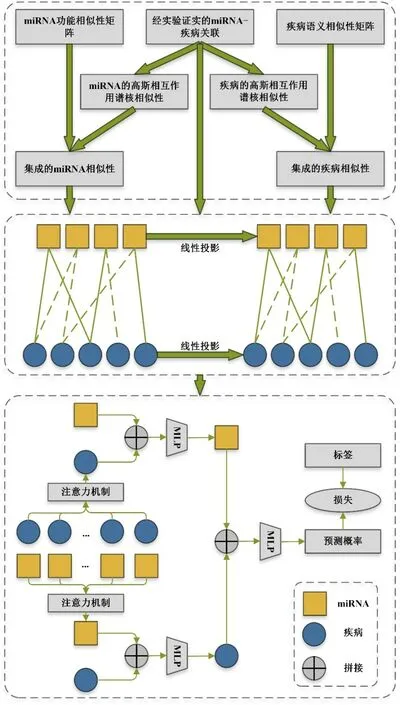
图1 基于异质图注意力网络的miRNA-疾病关联预测模型流程图
2 材料及方法
2.1 人类miRNA-疾病关联
本实验从“https://www.cuilab.cn/hmdd”下载了HMDD v2.0 和HMDD v3.0 数据集来对模型的预测效果进行验证[12]. 如表1 所示,经过数据预处理,HMDD v2.0 数据集中包含383 种疾病与495 种miRNA 间5 430条经实验证实的miRNA-疾病关联,HMDD v3.0 数据集中包含850 种疾病与1 057 种miRNA 间32 226 条经实验证实的miRNA-疾病关联. 为了便于存储,本实验采用二值矩阵A(nd×nm)来表示miRNA与疾病间的关联,其中nd 表示疾病数目,nm 表示miRNA 数目. 若疾病d(i)与miRNAm(j)有关联,则二值矩阵A对应位置的元素A(d(i),m(j))被赋值为1,否则为0.

表1 本文所用miRNA-疾病关联信息
2.2 MiRNA功能相似性
基于表型相似的疾病可能与功能相似的miRNA发生关联这一基本生物学假设,Wang 等人提出一种计算miRNA 功能相似性的模型[13]. 本实验从“https://www. cuilab.cn/files/images/cuilab/misim.zip”下载了miRNA 功能相似性数据,并构建出长度为nm的方阵FSM来存储miRNA的功能相似性.
2.3 疾病语义相似性
本实验基于美国国家医学图书馆的MeSH(Medi⁃cal Subject Headings)数据库计算疾病的语义相似性[14]. 疾病间抽象出的数据结构可以用有向无环图(Directed Acyclic Graph,DAG)进行表示. 具体而言,采用DAG(d(i)) =(d(i),T(d(i)),E(d(i)))来描述疾病d(i),其中,T(d(i))表示包含顶点d(i)自身及其祖先顶点的集合,E(d(i))表示包含从d(i)的祖先顶点到顶点d(i)的路径上所有直连的边的集合. 因此,疾病d(k)对d(i)的语义贡献值计算如下:
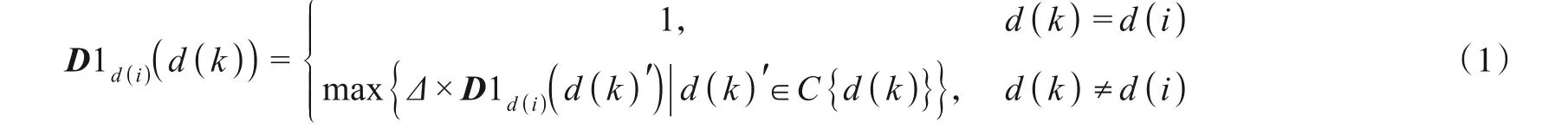
式(1)中,Δ表示语义贡献衰减因子,设置为0.5;C{d(k)}表示疾病d(k)的孩子顶点集合. 于是,疾病d(i)的语义值定义为

基于不同疾病间共享的DAG 部分越多,就具有更高的语义相似性这一假设(其中共享的DAG 部分指不同疾病顶点的祖先顶点的交集),疾病语义相似性矩阵DSSM1计算如下:
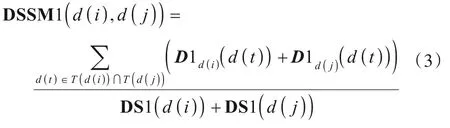
由于不同疾病在DAG 中出现的次数不尽相同,同一层DAG 中的疾病往往也会有不同的疾病语义贡献值,因此,根据疾病在DAG 中出现的次数计算另一种疾病d(k)对d(i)的语义贡献值的计算如下:

相应地,第二种疾病d(i)的语义值以及疾病的语义相似性矩阵DSSM2计算如下:
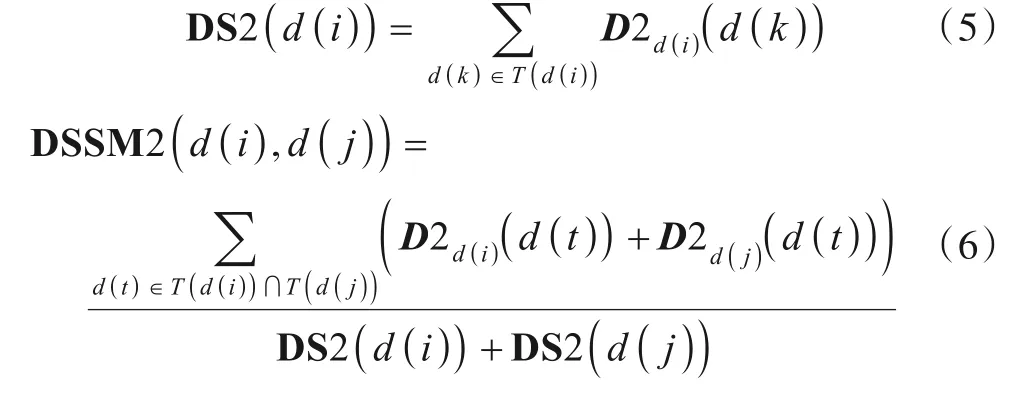
整合上述两种疾病语义相似性矩阵,计算最终的疾病语义相似性矩阵DSSM如下:

2.4 MiRNA与疾病的高斯相互作用谱核相似性
鉴于上述方法得出的miRNA 功能相似性矩阵以及疾病语义相似性矩阵具有稀疏性,本实验引入高斯相互作用谱核相似性[15]来进一步完善miRNA 和疾病的相似性信息. 根据miRNAm(i)是否与每一种疾病发生关联,构建二值向量IP(m(i))表示miRNA 的相互作用谱.miRNA的高斯相互作用谱核相似性矩阵MGSM为
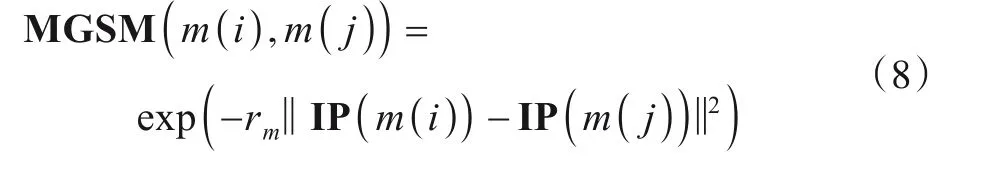
式(8)中,rm用以调控函数的带宽,可通过规范化参数计算而得:

式(9)中,r'm设置为1. 同样地,疾病的高斯相互作用谱核相似性矩阵DGSM可由下式计算:

其中,二值向量IP(d(i))表示疾病d(i)是否与每一种miRNA存在关联,r'd设置为1.
2.5 MiRNA与疾病的集成相似性
本文将miRNA 与疾病的高斯相互作用谱核相似性矩阵整合进miRNA 的功能相似性矩阵和疾病的语义相似性矩阵中,从而得到集成的miRNA相似性矩阵IM与集成的疾病相似性矩阵ID.

2.6 MiRNA-疾病异质图
本文构建了miRNA-疾病异质图,共包含两类顶点(分别为miRNA 顶点与疾病顶点),以及一类边(表示miRNA 与疾病间的关联). 其中,miRNA 顶点数目为nm,疾病顶点数目为nd,miRNA 与疾病间的关联数目为2S. 由于HMDD 数据集中经实验证实的miRNA-疾病关联数目远小于miRNA 与疾病间的未知关联数目,因此,从所有的未知关联中随机选取S条miRNA-疾病关联作为负样本. 在miRNA 和疾病顶点间相应地添加S条正边与S条负边,并将miRNA 的集成相似性信息赋给miRNAm(i)顶点,作为其属性特征Fm(i),即

2.7 异质图注意力网络
由于miRNA-疾病异质图中的miRNA 顶点和疾病顶点分别处于不同的特征空间中,对于每一种类型的顶点(例如类型为Φi的顶点),本实验设计了顶点类型转换矩阵WΦi将miRNA 顶点和疾病顶点投影到同一向量空间中进行计算,即

式(6)中,Fi和Hi分别表示顶点i的初始属性特征和投影后的属性特征;WΦi表示针对类型为Φi的顶点的投影矩阵,该矩阵可将不同向量空间的顶点投影至D维的向量空间中. 因此,miRNA顶点和疾病顶点可处在同一个向量空间中进行后续计算. 由于异质邻居顶点对中心顶点存在不同程度的影响,本实验采用多头注意力机制[16,17]聚合异质顶点的邻域信息,并将其与中心顶点的属性信息进行融合,从而得到包含异质图结构与顶点属性信息的miRNA 和疾病的有效特征嵌入. 首先计算中心顶点i与其邻居顶点j之间的注意力分数eij:

式(17)中,LeakyReLU 为非线性激活函数(负输入斜率为0.2). 仅计算顶点j∊Ni的注意力分数eij,其中,Ni表示顶点i的一阶异质邻居顶点集合. 采用softmax 函数规范化注意力分数eij,并计算出注意力权重系数αij,即
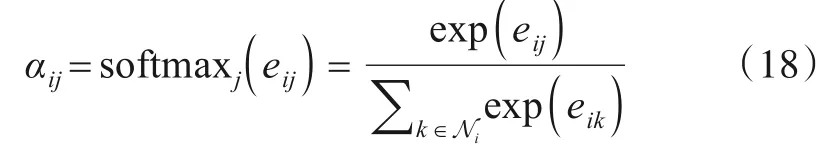
再根据顶点i的投影特征和注意力权重系数计算出顶点i的异质邻居聚合特征H'i,即

式(19)中,σ(∙)表示ELU 激活函数. 为了使模型学习到的特征嵌入更加稳定,按照上述公式独立计算K次,并将每次计算的结果拼接起来作为顶点i最终的异质邻居聚合特征H'i,即

上述过程仅聚合了异质邻居特征,却忽略了中心顶点特征,因此将异质邻居聚合特征H'i与中心顶点特征Fi拼接,并通过全连接层进行特征融合,表示为
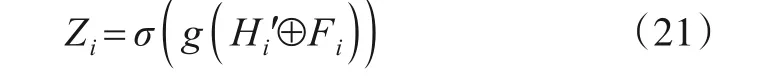
式(21)中,g(∙)表示输出维度为64的全连接层,⊕表示特征拼接操作. 最终分别获得64维度的miRNA 嵌入特征Zm和64维度的疾病嵌入特征Zd.
2.8 目标优化
为了获得miRNAm(i)与疾病d(j)间关联的预测概率,将上述得到的miRNA 和疾病嵌入特征拼接,并通过全连接层生成预测概率,即
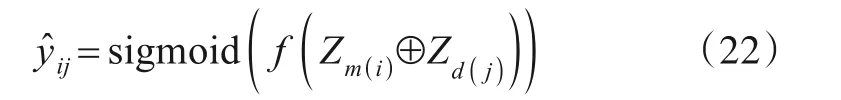
式(22)中,f(∙)表示输入维度为128,输出维度为1 的全连接层;sigmoid(∙)表示非线性激活函数.
本文采用交叉熵损失计算模型的预测值与标签间的损失,表示为

式(23)中,yij表示miRNAm(i)与疾病d(j)间的关联标签;Y和Y-分别表示正样本和负样本对应的顶点集. 最后,采用反向传播算法对整个模型进行端到端的训练.
3 实验结果与分析
3.1 实现细节
本实验基于深度图库(Deep Graph Library,DGL)[18]实现,后端采用PyTorch 框架,并采用Adam 作为模型的优化器. 经过网格搜索,设置学习率(Learning Rate)为0.0001,权重衰减(Weight Decay)为5×10-3. 为了防止过拟合,设置丢弃率(Dropout)为0.6. 为了保持较高的计算效率,设置多头注意力头数K为8,投影向量维度D为64. 为了充分训练模型的参数,训练批次(Epochs)设置为1 000.
3.2 评价指标
本文采用准确率(Accuracy)、精确率(Precision)、召回率(Recall)以及F1 值(F1-score)作为模型的评价指标,具体计算公式如下:

式(24)~(27)中,TP,TN,FP,FN 分别表示真正例数、真负例数、假正例数和假负例数. 此外,本文还绘制了受试者工作特征(Receiver Operating Characteristic,ROC)曲线以及精确率-召回率(Precision-Recall,P-R)曲线来直观地显示模型的预测能力,并分别计算了ROC 曲线下面积(Area Under the Curve,AUC)以及P-R 曲线下面积(Average Precision,AP)来综合评估模型的预测能力.
3.3 模型预测能力评估
本实验采用5 折交叉验证法(5-fold crossvalidation)对模型的预测能力进行评估. 本文所提模型在HMDD v2.0数据集上的预测结果如表2所示,取得了86.14%的准确率、86.10%的精确率、86.25%的召回率以及86.15%的F1 值. 所提模型在HMDD v3.0 数据集上的预测结果如表3 所示,取得了87.85%的准确率、88.02%的精确率、87.64%的召回率以及87.83%的F1值. 所提模型的5 折交叉验证ROC 曲线和P-R 曲线如图2 所示,该模型在HMDD v2.0 数据集上取得了93.52%的AUC 值和93.15%的AP 值,在HMDD v3.0 数据集上取得了94.82%的AUC 值和94.66%的AP 值. 由于HMDD v3.0 数据集中包含了更多的样本数量,且深度学习模型在更大的数据集上一般体现出更优的拟合效果,相较于HMDD v2.0 数据集,所提模型在HMDD v3.0数据集上关于6项评价指标均表现出更高的值. 为方便后续对比实验的展开,接下来的实验均采用HMDD v2.0数据集为基准数据集.

图2 所提模型基于5折交叉验证的实验结果图
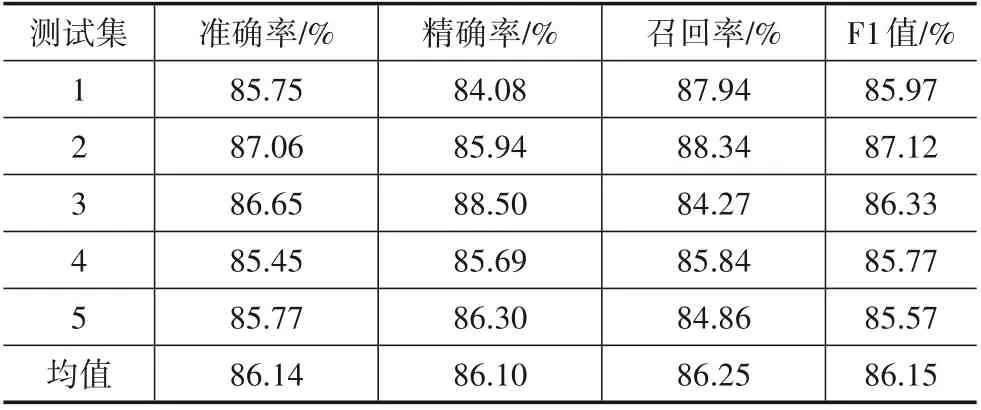
表2 所提模型基于5折交叉验证在HMDD v2.0数据集上的实验结果
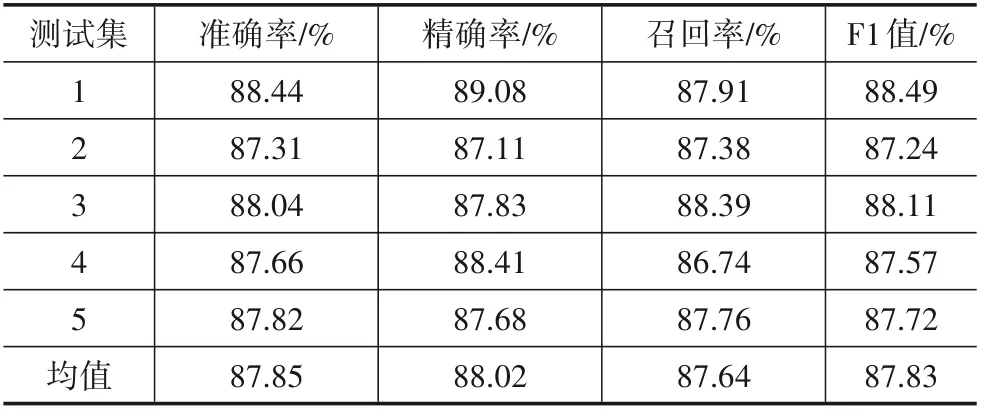
表3 所提模型基于5折交叉验证在HMDD v3.0数据集上的实验结果
3.4 特征融合的影响
本实验将中心顶点特征与其邻居聚合特征相融合作为最终的miRNA 和疾病的特征. 为了对比这种融合方式对模型预测能力的影响,本实验分别设计了只采用中心顶点特征的模型和只采用异质邻居聚合特征的模型,最终的对比结果如表4 所示. 从表中可以看出,本文所提模型在这三个模型中取得了最高的准确率、精确率、F1值、AUC 值以及AP值;尽管只采用邻居聚合特征的模型取得了最高的召回率,但其在其他5项指标上均远低于本文所提模型. 本文所提模型以多头注意力机制形式从多个角度探索miRNA-疾病异质图中异质顶点间复杂的交互信息,生成涵盖异质图结构及顶点属性信息的嵌入特征,进一步加强miRNA 和疾病特征的表达能力,提高模型的预测能力.
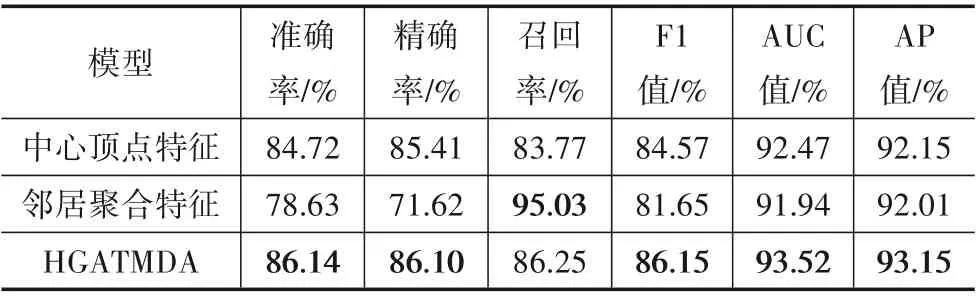
表4 所提模型与未进行特征融合的模型的对比实验结果
3.5 与其他方法的比较
为了进一步验证本文所提模型的有效性,将其与WBSMDA[19],BNPMDA[20],KBMFMDA[21],WBNPMD[22],M2GMDA[23],KNMBP[24],MCLPMDA[25]等7个模型基于5折交叉验证的平均AUC 值进行比较,此外,还对比了不同注意力头数K对所提模型AUC值的影响,详细的对比结果如表5 所示. 从表中可以看出,适当增加注意力的头数可以提高模型的预测能力,但过多的注意力头数反而会对模型预测能力起反作用. 最终,本文选择的注意力头数K=8,其对应的AUC值为93.52%,在所有8个模型中最高.
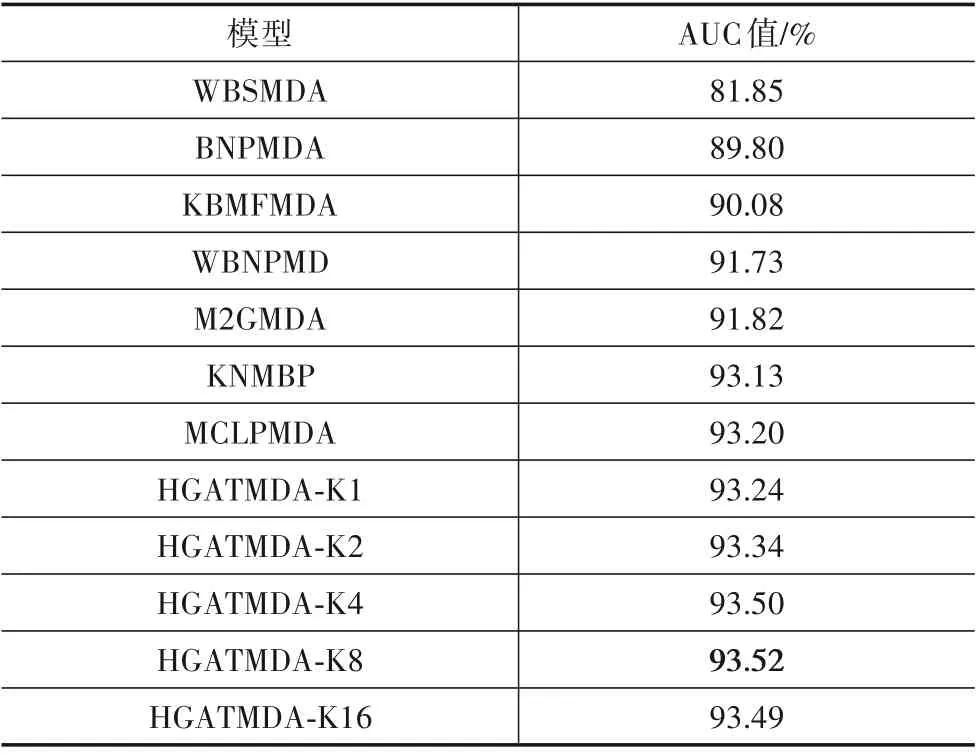
表5 所提模型与其他方法的AUC值的对比结果
3.6 病例研究
为了进一步评估本文所提模型在预测特定疾病潜在的相关miRNA 方面的性能,本文针对食管肿瘤疾病开展了病例研究. 首先采用HMDD v2.0 数据集对模型进行训练,然后预测与食管肿瘤有潜在相关的前50 种miRNA,最后通过dbDEMC[26]和miR2Disease[27]数据库进行验证.
食管肿瘤是一种发生在食管组织中的恶性肿瘤,全球范围内每年大约会有30 万人死于食管肿瘤. 本文选择食管肿瘤作为病例研究对象. 实验验证结果如表6所示,通过在dbDEMC 和miR2Disease 两个数据库中进行核实,模型预测的前25 个miRNA 中有24 个被证实,前50 个miRNA 中有48 个被证实. 因此,本文所提出的模型能有效预测出潜在的疾病相关miRNA,可作为一种便捷的工具指引研究人员开展相关具体的生物实验研究.
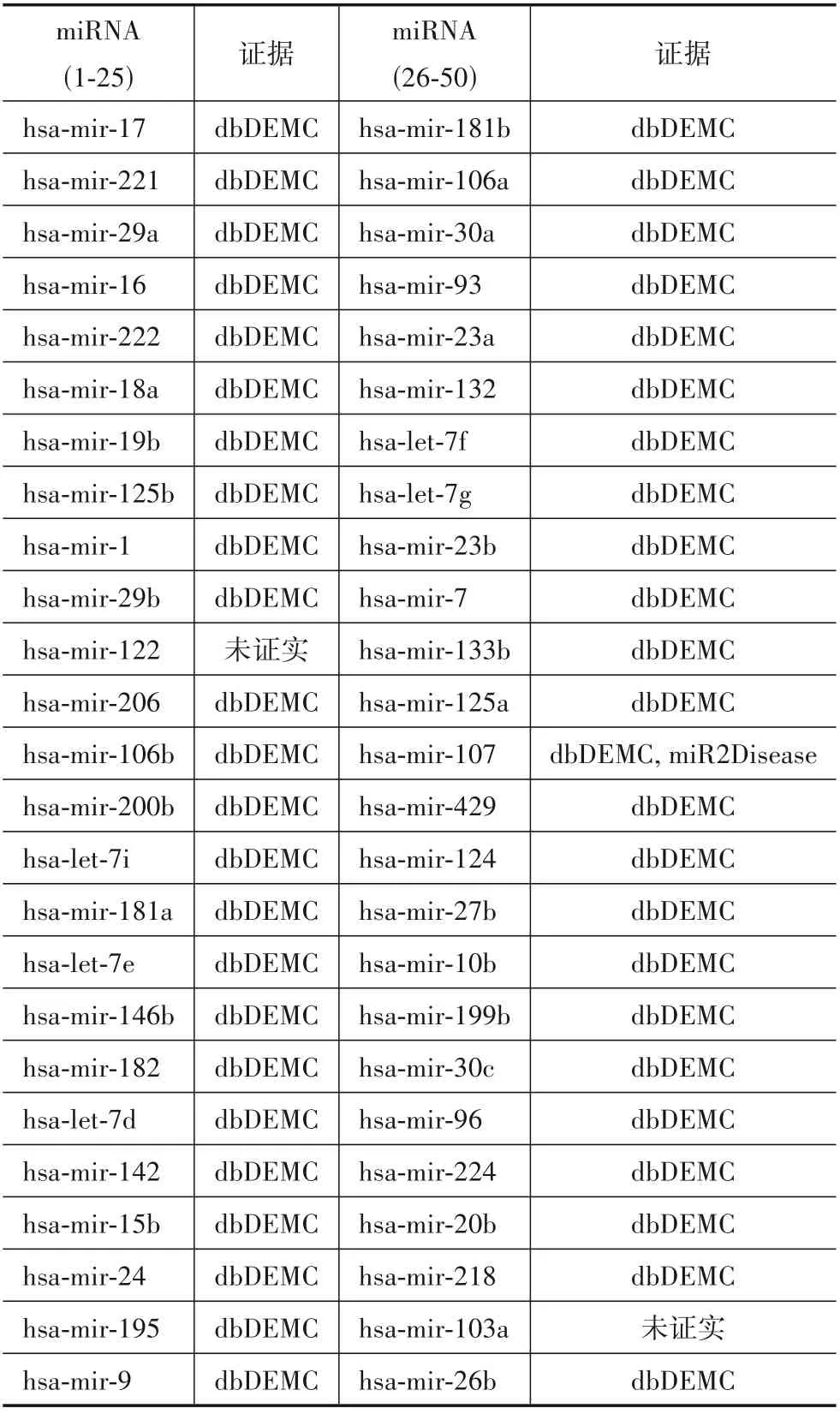
表6 所提模型预测出的前50个与食管肿瘤有关联的miRNA
4 结论
本文提出了一种基于异质图注意力网络的端到端计算模型(HGATMDA)预测潜在的miRNA 与疾病间的关联. 该模型首先将miRNA 和疾病间的多重相似性信息建模为异质图,并设计了顶点类型的转换矩阵将异质的顶点特征投影至同一向量空间中;然后采用多头注意力机制聚合中心顶点的异质邻居特征,并将其与中心顶点的特征进行有效融合,得到更具有表达能力的miRNA 和疾病特征嵌入;最后,将得到的miRNA 和疾病特征嵌入输入至全连接层中对潜在的miRNA 与疾病间关联进行预测.5 折交叉验证的结果表明,本文所提模型在多项评价指标上均取得了较为满意的结果.与未进行特征融合的模型的对比发现,本文所提模型的特征融合策略能够有效提升模型的预测性能. 此外,对食管肿瘤的病例研究结果也显示出所提模型具有良好的预测能力. 上述实验结果均表明,本文提出的计算模型可作为预测miRNA 与疾病间潜在关联的可靠工具. 在接下来的研究中,将尝试在模型中嵌入更多的多源信息,如miRNA 序列信息、靶基因信息等,以期进一步提升模型的预测性能.
猜你喜欢
数学物理学报(2022年5期)2022-10-09
家庭教育报·教师论坛(2021年42期)2021-12-23
中等数学(2021年9期)2021-11-22
中等数学(2021年8期)2021-11-22
河北画报(2021年2期)2021-05-25
北方论丛(2021年2期)2021-05-22
河北画报(2020年8期)2020-10-27
环球市场信息导报(2017年1期)2017-04-08
公务员文萃(2013年5期)2013-03-11
俄罗斯问题研究(2013年1期)2013-03-11
