
尿酸作用于Keap1/Nrf2通路调控强直性脊柱炎氧化应激的研究进展
2022-06-23 12:58邬秀娣
临床荟萃 2022年5期
俞 杭,邬秀娣
(1. 宁波大学医学院,浙江 宁波 315211;2. 宁波市第一医院 风湿免疫科,浙江 宁波 315000)
强直性脊柱炎(ankylosing spondylitis,AS)是一种免疫介导的慢性炎症性疾病,通常影响中轴及外周关节,是青壮年男性致残的主要疾病之一[1]。AS 最常见的关节外表现包括炎症性肠病(高达50%)、急性前葡萄膜炎(25%~35%)和银屑病(约10%)[2]。AS不仅给患者带来了躯体痛苦,也给社会和家庭增加了经济负担[3]。
AS患者的预后与早诊断早治疗密切相关。X射线是用于评估AS患者骨盆和脊柱关节改变最广泛使用的诊断和监测方法,但AS患者进展至关节放射学异常表现往往需要几年时间[4]。MRI可发现AS的早期炎症性改变[5],有助于早期诊断AS,但成本高、适用范围小[6],不适合短期内反复检查以评估病情。目前AS的疾病活动性评估往往依赖血沉(ESR)和C反应蛋白(CRP)这些炎症标志物,但并不总是能精准反映AS疾病的活动性和进展[7]。故还需要寻找其他指标以早期发现并联合评估AS的疾病活动性。
AS的病因和发病机制尚未完全阐明,目前认为它涉及炎症性骨侵蚀和异常的新骨形成[8]。越来越多的学者发现AS患者的血清尿酸水平异常变化,研究认为尿酸与机体的氧化应激相关[9]。信号通路Kelch样ECH相关蛋白1(Kelch-like ECH-associating protein 1, Keap1)-NFE2相关因子2(NFE2 related factor 2, Nrf2)参与各种病理过程中的细胞保护机制,被认为是氧化应激的敏感传感器和调节器[10]。已有研究认为尿酸通过调节Keap1/Nrf2参与了AS的氧化应激调控,但其中的具体关系还存在着一些争议,故本文就相关研究进展做一综述。
1 AS患者疾病活动性与尿酸的关系
AS患者活动期主要表现为晨僵、夜间腰背痛和外周关节炎症[11], Zhu等[12]通过双能计算机断层扫描技术检测到大量尿酸单钠晶体沉积在不伴有痛风的中轴型脊柱炎患者的骨盆处,且这些患者骶髂关节的尿酸单钠晶体沉积与其影像学分级进展相关,提示尿酸盐沉积可能参与了AS患者的关节炎症。
Zhang等[13]测定94名AS患者的尿酸水平后显示23.4% AS患者合并高尿酸血症,且与正常尿酸水平患者相比,该类患者肝肾功能较差,AS疾病活动度却更低。而曾沛英等[14]将362例AS患者的临床数据分析后则显示AS合并高尿酸血症组晨僵时间更短、IgM水平更低、ESR更低,与尿酸正常组相比有更低的病情活动度和更少的并发症。这些都提示AS与尿酸代谢存在一定关系,而血尿酸水平与AS疾病活动度的确切关系值得进一步探索。
2 AS发病机制与尿酸的关系
2.1尿酸作用于Keap1/Nrf2通路激活氧化应激 AS患者具有病态的血清微环境和较强的氧化应激,长期暴露于氧化环境通常会导致细胞产生过量的活性氧,诱导细胞衰老并伴有细胞功能障碍[15]。Dong等[16]以小鼠为研究对象,发现了BALB/c小鼠的氧化指标丙二醛水平高于健康小鼠,而抗氧化酶、过氧化氢酶和谷胱甘肽过氧化物酶水平等抗氧化指标则比健康小鼠低。
Nrf2调节编码可诱导细胞保护蛋白的大型基因网络的表达,使哺乳动物细胞和生物体能够在包括氧化应激在内的各种条件下适应和生存。Nrf2与其主要的负调节因子Keap1一起,形成了一个分子效应器和传感器系统,对细胞氧化还原平衡的干扰做出强有力的反应,并协调一个全面的防御计划,从而恢复内环境平衡[17]。在生理环境中,Nrf2位于细胞质中,并与控制Nrf2活性的Keap1结合。氧化或电应激诱导Keap1的构象变化或直接促进Nrf2的磷酸化,促使Nrf2从Keap1中分离出来,并转移到细胞核,与抗氧化反应元件(antioxidant-responsive element, ARE)进行结合[18]。
另有研究证实尿酸降低了Nrf2的泛素化和降解,促进了其核易位,推动了Nrf2靶向的抗氧化基因的转录和翻译[19]。一般认为,尿酸激活Nrf2可能是由于Nrf2与Keap1分离,促使Nrf2从Keap1介导的蛋白酶体降解中逃逸。从结构上看,尿酸呈酮烯醇互变异构体形式,可与Keap1的半胱氨酸残基反应,阻止泛素连接酶将Keap1和Nrf2结合[20]。这些结果都有力地证明了尿酸激活Nrf2信号通路的抗氧化作用。
但是对尿酸调控Keap1/Nrf2方面也有学者有不同的见解。沈瑞明等[21]的研究分析显示尿酸通过活化NADPH氧化酶依赖性途径产生细胞内氧化物,导致AS患者活性氧、活性氮、丙二醛水平的升高,进一步促进了Keap1的表达;而Keap1与Nrf2结合之后会导致Nrf2的表达水平降低,Keap1/Nrf2信号通路被抑制,减弱机体的抗氧化能力,进而加重AS患者的炎症反应,见图1。
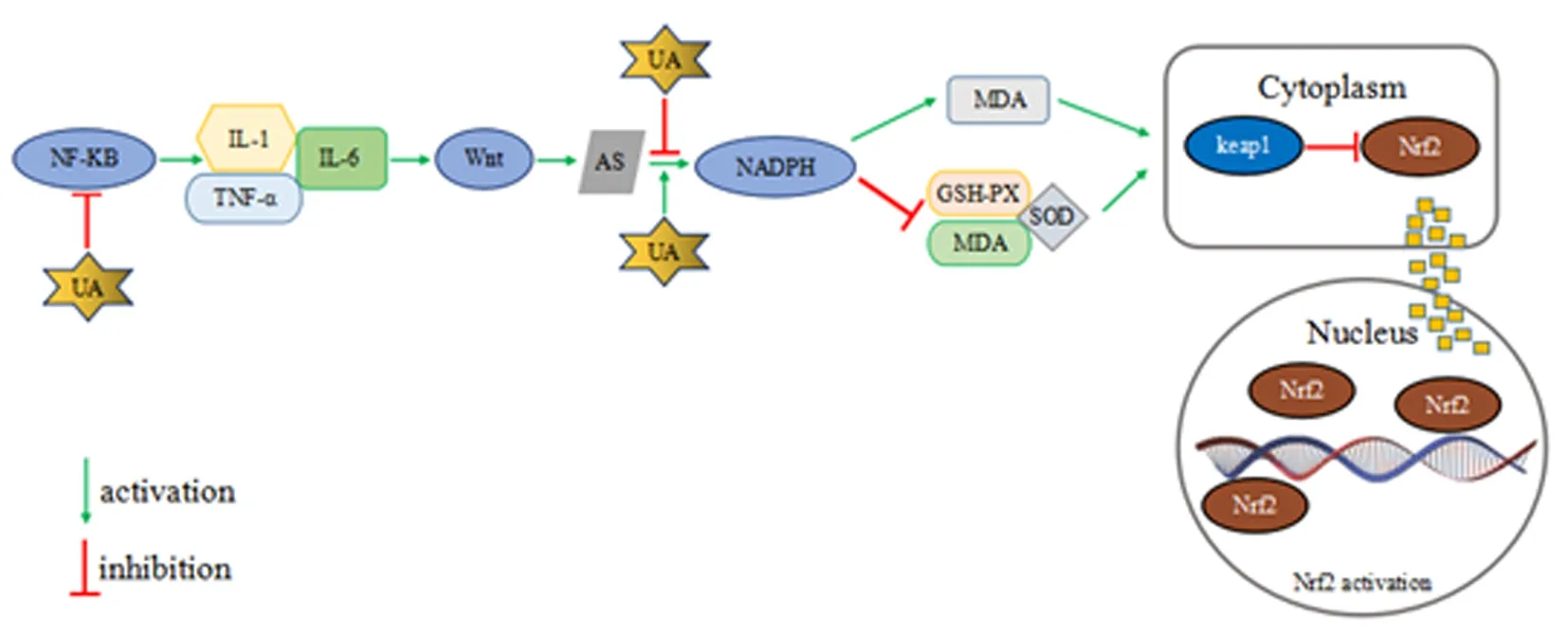
图1 尿酸作用于Keap1/Nrf2通路调控AS氧化应激的机制
2.2尿酸与AS患者靶器官损伤的关系
2.2.1尿酸影响AS患者的骨代谢 AS主要发生于脊柱、骨骼、外周关节和关节外组织,在发病初期,50%~92%的AS患者伴有骨质减少或骨质疏松[22]。最近的研究发现,骨代谢的生化变化先于全身性骨质疏松症和关节僵硬[23]。
氧化应激是骨质疏松症的病理机制之一。核因子κB(nuclear factor κB,NF-κB)是一种蛋白质复合物,存在于细胞质中,活化的NF-κB能够促进多种细胞因子基因的表达,如白细胞介素(IL)-1、IL-6和肿瘤坏死因子(TNF)-α[24]。已证实,TNF-α和IL-6是AS致病机制中重要的炎症因子,均可在刺激破骨细胞的同时抑制成骨细胞的生成[25]。尿酸能够作用于NF-κB通路,下调炎症因子的表达,从而预防细胞损伤[26]。有学者结合实验结果确定尿酸通过减少活性氧来抑制破骨细胞形成,再次验证了尿酸与骨密度之间的正相关与尿酸的抗氧化作用有关,即尿酸对骨代谢有保护作用[27]。活性氧在调节NF-κB配体(RANKL)依赖性破骨细胞分化的受体激活过程中充当细胞内信号分子,但它们也具有细胞毒性作用,包括脂质过氧化和对蛋白质和DNA的氧化损伤[28]。用RANKL刺激破骨细胞前体能够上调Keap1,并且降低Nrf2/Keap1的比率[28]。Nrf2缺失或过度激活均会导致小鼠的骨质明显减少,且 Keap1-/-成骨细胞的增殖潜力明显低于 Keap1+/-成骨细胞[29]。
结合临床分析,一项大型横断面研究表明高尿酸血症的绝经后女性有着更丰富的脂肪含量,有益于骨骼健康[30]。对2 981例卡塔尔成年人进行横断面研究,调整年龄和性别后分析得出相对高的尿酸水平(>200 μmol/L)与全身和特定部位骨骼位置的骨矿物质密度增加显著相关[31]。有学者研究发现,血清尿酸浓度与骨吸收标记物I型胶原末端肽的尿排泄呈负相关[32]。尿酸与AS骨代谢的关系也值得进一步的深入探讨。
2.2.2尿酸影响AS患者肾脏受累 研究均表明,AS患者往往伴有血清尿酸升高[12-13]。Wu等[33]对926例AS患者行回归分析后提示尿酸水平的增加可增加AS患者发生肾脏受累的风险,一旦发生肾脏受累,会严重影响AS患者的预后,相应的死亡率为4%。尿酸水平升高和肾脏受累之间会相互促进,肾动脉硬化和肾小球肾炎均可导致肾小球滤过和肾小管分泌减少,从而导致尿酸水平升高[33]。在AS早期,炎症因子CRP和ESR会迅速升高[34],这也与肾功能恶化密切相关[33]。
AS最常见的病理类型是IgA肾病,其诊断主要依靠肾活检[35]。Zhang等[35]将71例继发性IgA肾炎的AS患者纳入分析,发现HLA-B27阴性患者在基线时的尿酸水平明显高于HLA-B27阳性患者;病理上,在HLA-B27阴性患者中观察到患者的整体肾小球硬化比例更高,在存在大量蛋白尿或间质纤维化和肾小管萎缩的患者的亚组分析中也发现了这种显著关联。这些提示HLA-B27基因可能与尿酸代谢存在一定联系,而尿酸水平又影响着AS患者的肾脏累及及预后。
在机制研究方面,氧化应激被认为是各种进行性临床和实验型肾脏疾病的主要致病因素[36]。Nrf2具有抗炎特性,其主要通过细胞保护基因表达的转录激活来发挥其抗氧化功能[37]。Nrf2的激活能够清除毒素和活性氧[38],改善各种肾脏疾病中的氧化损伤和炎症,作用于Nrf2抗氧化途径是肾脏疾病治疗的重要靶点[39]。研究证明尿酸可促进Nrf2向细胞核的移位并减少Nrf2泛素化[40],但它并不影响Keap1蛋白的表达。越来越多的证据支持Nrf2在保护肾脏抵抗多种损伤方面发挥重要的生理作用[41]。
2.2.3尿酸和AS心血管内皮改变相关 在免疫介导的疾病中,慢性炎症和免疫失衡机制被认为与加速动脉粥样硬化有关[42]。内皮功能障碍作为动脉粥样硬化序列中的首发事件,通过早期量化颈总动脉的硬化指数发现34.29%的AS患者有动脉粥样硬化,而对照组仅为5.71%[43]。而AS作为一种典型的慢性炎症性疾病,其动脉粥样硬化的发生与尿酸的关系也值得进一步探索。
活性氧的产生是动脉粥样硬化过程中的中心致病因素,活性氧可以诱导低密度脂蛋白氧化为氧化型低密度脂蛋白(oxLDL),并且oxLDL在动脉壁中的积累会导致促炎事件,包括巨噬细胞和淋巴细胞的募集,从而引发随后的动脉粥样硬化病变形成[44]。在小鼠模型中,Nrf2表达量的增加通过Ⅱ期抗氧化酶活性间接保护巨噬细胞免受oxLDL介导的损伤[45]。Nrf2的下游靶基因HO-1能够通过减少oxLDL诱导的单核细胞迁移来抑制动脉粥样硬化病变的形成[46]。最新研究证实尿酸激活了Nrf2抗氧化途径,并对氧化低密度脂蛋白诱导的内皮损伤具有保护作用[40]。
血尿酸通过内皮细胞氧化应激和糖萼脱落诱导内皮细胞向间充质细胞的转化[47]。正常血清尿酸浓度可能具有抗氧化作用,但血液中过饱和状态的尿酸可能通过增加活性氧的产生,从而加强机体的氧化应激[48]。在无症状高尿酸血症患者的多个关节和身体软组织中也能够观察到尿酸单钠晶体的沉积,并与冠状动脉钙化的严重程度增加相关[49-50]。
3 AS尿酸浓度监测的临床意义
韩国一项包括150例AS患者的研究表明,尿酸水平与AS患者的骨密度呈正相关。随着尿酸水平的增加,骨密度水平增加[51]。孙文婷等[52]收集分析了143例AS患者的尿酸数据后得出关于血尿酸水平和腰椎股骨粗隆骨量变化之间的一个线性关系,即血尿酸水平在150~600 μmol/L时,血尿酸水平越高,骨量减少的风险就越低。在氧化应激的早期,抗氧化酶的活性可能会增强,以消除细胞内活性氧的沉积。然而,当细胞内活性氧积累不能被抗氧化系统中和时,则会引发氧化损伤,抗氧化酶的活性反而减弱[53]。
黎荣山等[54]将柳州2 150例AS患者的数据纳入回顾性研究,研究发现维持一定水平的尿酸可以预防骨质流失,但是,过高浓度尿酸则伴骨密度降低。Chen等[55]将182例AS患者(其中包括143例男性和39例女性患者)纳入研究,根据尿酸数值将患者分为3组:<300 μmol/L;300~360 μmol/L,>360 μmol/L组。结果显示300~360 μmol/L组内的尿酸患者骨密度明显高于其他2组,观察到AS患者的尿酸与骨密度之间的关系为倒“U”型,保持尿酸水平在300~360 μmol/L范围内有助于减少AS患者的骨质疏松症发病率,并进一步减少由骨质疏松导致的骨折及相应的严重并发症。另外, Zhang等[56]研究显示适当浓度的尿酸(300 μmol/L)可以显著增加神经元的活性,并减少活性氧的产生。Lin等[40]设立1、2、3、5、6、9、12、15和18 mg/dl梯度的尿酸浓度进行实验,结果发现0~5 mg/dl尿酸不会诱导内皮细胞损伤,5 mg/dl尿酸可以显著减少血管内皮损伤,>5 mg/dl的尿酸与氧化低密度脂蛋白对内皮细胞的损伤有协同作用。这与以上研究者的结果存在矛盾,也提示定量确认AS患者与合理尿酸水平之间的关系还需要进一步大样本的数据分析。
4 展望
AS作为严重危害青壮年人群健康的常见慢性炎症性疾病,其发病机制复杂。根据已有的研究结果显示,尿酸水平的变化可能作用于Keap1/Nrf2通路调控AS氧化应激。但是目前的研究仍存在一些局限性:①尿酸在AS患者中具体是上调还是抑制Nrf2的表达存在争议,未达成共识。②临床研究病例样本小,研究时段不够长,覆盖范围不够广。③目前对于AS患者的尿酸水平定量化研究仍相对较少,严格把控尿酸水平在一个定值目前较为困难。故进一步明确血尿酸水平与AS的关系,并探索将血尿酸水平监测运用于AS患者病情与预后评估及治疗选择,尚需要深入研究和探讨。
猜你喜欢
临床肝胆病杂志(2022年10期)2022-10-19
茶道(2022年3期)2022-04-27
家庭科学·新健康(2022年2期)2022-03-07
科学导报(2022年11期)2022-03-03
老年世界(2018年2期)2018-05-21
食品与健康(2017年4期)2017-04-20
江苏农业科学(2016年8期)2017-02-15
风湿病与关节炎(2016年12期)2017-01-14
科学与管理(2016年5期)2016-12-01
中国中药杂志(2016年20期)2016-11-19
