
新生儿常见小耳畸形相关综合征的遗传特征
2022-06-22 07:28:34马竞周文浩
中国当代儿科杂志 2022年6期
马竞 周文浩
(1.复旦大学附属眼耳鼻喉科医院眼耳鼻整形外科,上海 200031;2.复旦大学附属儿科医院新生儿科,上海 201102)
小耳畸形(microtia)又称先天性外中耳畸形,是一种以耳廓位置、大小、形态和结构异常为主要表现的常见新生儿先天性畸形,多合并外耳道狭窄或闭锁及多种中耳畸形,是仅次于唇腭裂的颌面部第二大出生缺陷。全球发病率为0.8/10 000~17.4/10 000[1],尤其在美洲、北欧及亚洲发病率较高,我国的发病率为3.06/10 000[2]。
外、中耳由内胚层的第一咽囊,外胚层的第一、第二鳃弓及中胚层间充质发育而来[3]。由于遗传因素在各胚层的发育过程中都发挥了重要作用,胚胎发育早期的遗传变异会导致包括外中耳在内的各胚层来源的多器官受累。因此,临床上有20%~60%的小耳畸形是作为某种类型综合征的耳部症状出现[1]。目前已报道的能够引起耳畸形的综合征已达90余种,绝大多数都是单基因遗传病,其中发病率前10位的小耳畸形相关综合征(microtia-associated syndromes,MAS)总结见表1。其中发病率高、耳畸形发生率高(>80%)且耳畸形表型严重及听力受损严重的3大综合征分别是眼-耳-脊椎畸形谱系(oculo-auriculo-vertebralspectrum,OAVS)、鳃-耳-肾谱系疾病(branchiooto-renal spectrum disorder,BORSD)及Treacher Collins综合征(Treacher Collins syndrome,TCS),本文将重点论述这3种MAS的临床表型和遗传学特征,并概述其他3种相对常见、耳畸形发生率较高但听力常不受损的综合征:22q11.2微缺失综合征(22q11.2 deletion syndrome,22q11.2 DS)、Noonan综合征(Noonan syndrome,NS)及CHARGE综合征(CHARGE syndrome,CS)。
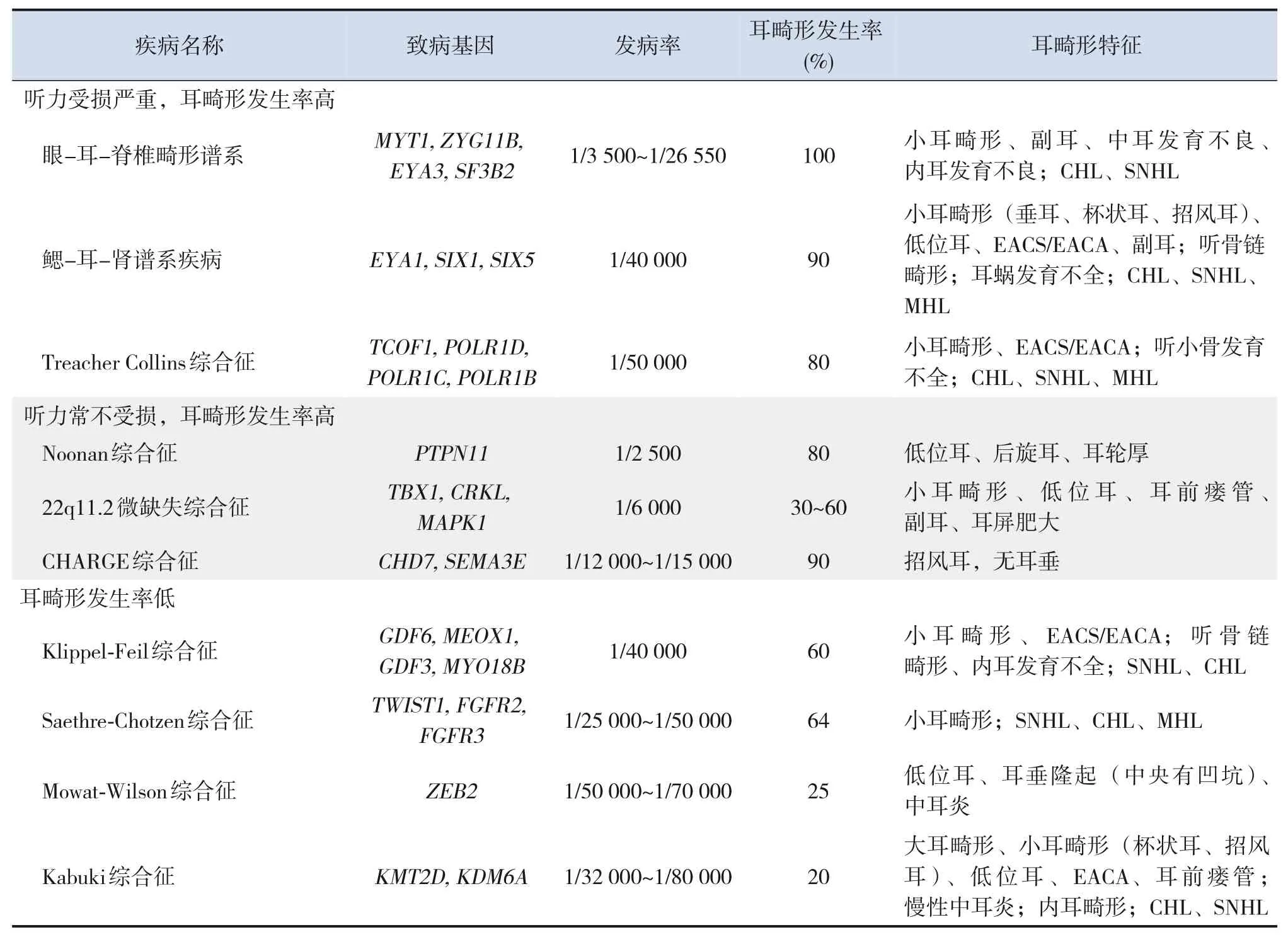
表1 发病率前10位的MAS[4-5]
1 OAVS
1.1 概述
OAVS又称半面短小畸形,第一、第二鳃弓综合征,眼-耳-脊柱综合征或Goldenhar综合征等,是以眼、耳、面部及脊柱畸形为主要特征的一类遗传病的统称[6]。
1.2 临床表型
眼部畸形以眼部表皮样囊肿为主。耳畸形包括小耳畸形、副耳、耳前瘘管、听骨链畸形及中耳腔缩小等,内耳畸形较少见。面部异常还包括单侧上/下颌骨发育不全致面部不对称(半面短小)、巨口、唇腭裂、面瘫、眼外肌及三叉神经麻痹。脊椎畸形包括脊柱侧弯、骨质融合、脊柱裂等。
其他临床表现包括先天性心脏病、泌尿生殖道畸形、脑发育不全、食管闭锁、肺发育不全等。小眼畸形患者常伴有智力障碍。
1.3 遗传学病因
OAVS呈常染色体显性(autosomal dominant,AD)或常染色体隐性(autosomal recessive,AR)模式遗传,目前明确的致病基因有:
髓鞘转录因子1(myelin transcription factor 1,MYT1)基因定位于染色体20q13.33,含23个外显子。该转录因子与中枢神经系统脂蛋白的编码基因启动子区域结合,参与神经系统发育。2016年首次在OAVS患者中鉴定出MYT1基因突变,敲除myt1a基因的斑马鱼出现颅面软骨畸形。体外功能研究显示MYT1基因过表达和突变会影响视黄酸受体基因的表达,干扰视黄酸通路,导致OAVS相关的表型[7]。
Zyg-11家族成员B(zyg-11 family member B,ZYG11B)基因编码的蛋白质是一种细胞周期调节因子。ZYG11B基因定位于染色体1p32.3,含16个外显子。2020年首次在OAVS患者中鉴定出ZYG11B基因突变,敲除zyg11基因的斑马鱼出现颅面部软骨发育不良及小眼畸形[8]。
眼缺乏同源物3(eyes absent homolog 3,EYA3)基因编码的转录共激活因子参与胚胎发育过程。EYA3基因定位于染色体1p35.3,含有22个外显子。2021年首次在OAVS患者中鉴定出突变,体外实验证实突变造成EYA3蛋白半衰期延长,eya3基因敲除的斑马鱼出现颅面部发育障碍[9]。
剪切因子3B亚基2(splicing factor 3b subunit 2,SF3B2)基因编码的蛋白质是U2小核糖核蛋白复合物组成部分。SF3B2基因定位于染色体11q13.1,含22个外显子。2021年在OAVS患者中发现SF3B2基因突变,敲除SF3B2基因的非洲爪蟾出现颅神经嵴细胞异常及颅面软骨畸形[10]。
2 BORSD
2.1 概述
BORSD是胚胎期鳃器、耳及肾脏发育异常引起的鳃裂、耳及肾脏畸形为主的疾病。BORSD包括伴肾脏异常的鳃-耳-肾综合征(branchio-otorenal syndrome,BOR)及不伴肾脏异常的鳃-耳综合征(branchio-oto syndrome,BOS)。
2.2 临床表型
患者表现出多种结构畸形,以鳃裂、耳部、肾的异常为著。
鳃裂异常包括鳃裂瘘管、窦道或囊肿。外耳畸形包括垂耳、杯状耳、招风耳、低位耳等。中耳畸形包括听骨链畸形、错位、脱位或固定及中耳腔缩小等,患者可有传导性、感音神经性或混合性耳聋。肾异常表现为肾发育不全及肾缺如,也包括集合系统畸形及交叉性肾异位等。
其他异常包括泪道异常、腭裂、甲状腺肿、面神经麻痹、精神运动发育迟缓等。
2.3 遗传学病因
BORSD呈AD模式遗传,致病基因包括:
眼缺乏同源物1(eyes absent homolog 1,EYA1)基因编码的转录共激活因子有酪氨酸磷酸酶和转录辅助激活作用。EYA1基因定位于染色体8q13.3,含25个外显子。1997年首次在患者中鉴定EYA1基因突变并发现其与肾脏、鳃弓、耳发育的关系。体外研究发现EYA1基因突变可促进EYA1蛋白的蛋白酶体途径降解。果蝇eya基因突变导致其酪氨酸磷酸酶活性的丧失及激活转录能力的降低。eya1基因杂合敲除小鼠表现为传导性耳聋和肾脏异常[11]。
SIX同源框1(SIX homeobox 1,SIX1),又名常染色体显性遗传性耳聋23(deafness,autosomal dominant 23,DFNA23)基因,编码的转录因子介导EYA1的核转移,影响细胞周期及凋亡途径,参与耳发育。SIX1基因定位于染色体14q23.1,含2个外显子。2004年首次在患者中鉴定SIX1基因的突变,证实突变影响EYA1与SIX1的相互作用。six1基因敲除小鼠具有内耳、肾脏的发育缺陷。SIX1蛋白参与感觉发育早期的表达模式调控,SIX1基因突变可影响胚胎颅面基因表达及听囊发育[12]。
SIX同源框5(SIX homeobox 5,SIX5)基因编码的转录因子与SIX1基因同属SIX家族,参与调控器官发生及视网膜形成。SIX5基因定位于染色体19q13.32,含3个外显子。2007年首次在患者中鉴定SIX5基因的突变,突变影响SIX5蛋白及SIX5-EYA1蛋白复合体的转录激活作用[13]。
Meta分析显示,EYA1基因突变更易导致严重的外耳畸形及中重度混合性耳聋,SIX1基因突变更易导致重度感音神经性耳聋[14]。
3 TCS
3.1 概述
TCS是胚胎第一、第二鳃弓发育异常导致颅面畸形的先天性遗传病,引起颧骨、上颌骨、下颌骨等的发育不良,又称鸟面综合征。
3.2 临床表型
患者表现出多种结构畸形,以中下面部、眼部、耳部、口腔、呼吸道异常为著[15]。
中、下面部发育不良形成的面容俗称为“鸟面”,颧骨发育不全常伴两侧眼眶不对称、颊部的骨性突起消失。眼部常表现为小睑裂及睑裂下斜,亦可有下眼睑和虹膜的缺损、睑外翻、睑内侧睫毛缺如、泪腺发育不良。耳畸形包括外中耳畸形、低位耳。口腔异常包括腭裂、牙列不齐及咬合不良。
其他异常包括舌后坠、气道狭窄、鼻中隔偏曲、后鼻孔闭锁、颞下颌关节功能异常等。患者智力通常不受影响。
3.3 遗传学病因
TCS呈AD或AR模式遗传,致病基因总结如下:
Treacle核糖体生物发生因子1(treacle ribosome biogenesis factor 1,TCOF1)基因编码的Treacle蛋白是参与核糖体合成的一种核仁磷酸蛋白,与上游结合因子作用,参与由RNA聚合酶I催化的核糖体DNA的转录及转录后修饰。TCOF1基因定位于染色体5q32,含29个外显子。tcof1基因杂合突变导致小鼠神经嵴细胞的核糖体合成减少,细胞内源性凋亡通路激活,向颅面区域迁移的能力减弱,导致颅面畸形[16]。
RNA聚合酶Ⅰ和Ⅲ亚基D(RNA polymeraseⅠandⅢsubunit D,POLR1D)基因、RNA聚合酶Ⅰ和Ⅲ亚基C(RNA polymeraseⅠandⅢsubunit C,POLR1C)基因和RNA聚合酶Ⅰ和Ⅲ亚基B(RNA polymeraseⅠandⅢsubunit B,POLR1B)基因编码的蛋白为RNA聚合酶的组成部分,参与核糖体合成。POLR1D基因定位于染色体13q12,含6个外显子;POLR1C基因定位于6p21,含12个外显子;POLR1B基因定位于2q14.1,含18个外显子。POLR1D基因突变导致单倍体功能不全,POLR1C基因突变导致蛋白质功能缺失,引起胚胎发育关键时期神经上皮细胞及神经嵴细胞中的核糖体数量不足,激活细胞死亡通路,致颅面畸形。polr1c基因和polr1d基因的纯合突变可导致斑马鱼软骨发育不全和颅骨异常。polr1b基因敲除的斑马鱼存在RNA聚合酶Ⅰ特异性的核糖体RNA转录缺陷,出现颅面部发育异常[17-18]。
4 其他综合征
4.1 22q11.2 DS
22q11.2 DS是染色体22q11.21~22q11.23区域片段缺失引起的临床表现各异的一系列综合征的统称,包括以心脏畸形、免疫缺陷及低钙血症为主要表现的DiGeorge综合征(DiGeorge syndrome,DGS);以腭裂、面部畸形、心脏畸形及学习障碍为特点的腭心面综合征(velocardiofacial syndrome,VCFS);以心脏畸形、面部畸形为特征的椎干异常面容综合征(conotruncal anomaly face syndrome,CAFS)等。面部畸形的患者具有小耳、低位耳、耳前瘘管、副耳、耳屏肥大等耳畸形表型。位于22q11.2的致病基因包括T-box转录因子1(T-box transcription factor 1,TBX1)基因[19]、CRK样促癌(CRK like proto-oncogene,adaptor protein,CRKL)基因[20]及线粒体激活蛋白酶1(mitogen-activated protein kinase 1,MAPK1)基 因[21],以AD模式遗传。
4.2 NS
NS以倒三角形脸、低位后旋耳等特殊面容、先天性心脏病及身材矮小为特征。NS按不同的致病基因可以分为13型。其中,因蛋白酪氨酸磷酸酶非受体11型(protein tyrosine phosphatase nonreceptor type 11,PTPN11)基因导致AD模式遗传的NS1型患者最为常见,占50%[22]。因SOS Ras/Rac鸟嘌呤核苷酸交换因子1(SOS ras/rac guanine nucleotide exchange factor 1,SOS1)基因导致的NS4型患者占13%[23],也以AD模式遗传。其余致病基因占比均低于5%。
4.3 CS
CS表现为眼部缺损(coloboma,C)、先天性心脏病(heart disease,H)、后鼻孔闭锁(atresia of choanae,A)、精神及生长发育迟滞(retardation of mental and somatic development,R)、生殖器发育不 全(genital hypoplasia,G)、耳 异 常(ear anomalies,E)等。致病基因包括染色质域解旋酶DNA结合蛋白7(chromodomain helicase DNA binding protein 7,CHD7)基因[24]和人信号素3E(semaphorin 3E,SEMA3E)基因[25],以AD模式遗传。
5 结语
近年来,随着致病基因的确定及遗传学检测手段的发展,MAS的诊断率不断提高、表型谱也不断完善。但目前仍有许多病例无法用现有的致病基因解释,需要通过更多手段探索新致病基因。
当发现新生儿存在耳畸形,尤其是合并多发畸形的,都应进行基因检测。由于不同MAS间的表型相互交叉,且大部分MAS都存在不完全外显、表型高度可变的特点,同一家系中的不同患者的受累程度也各不相同,因此,基因检测是明确病因、辅助优生优育的唯一手段。由于高通量测序的数据庞大,使用表型-基因型分析软件对基因和疾病表型的相关性评分,提高数据分析的效率和准确性,有助于致病突变的高效筛选[26]。
由于MAS涉及的器官发育都于人类胚胎发育早期完成,产前诊断是预防此类出生缺陷的重要手段,但目前成熟的单基因疾病产前诊断方法均有创伤性。非侵入性产前检测(noninvasive prenatal testing,NIPT)可通过取材母体血浆游离DNA,结合二代测序、相对单倍型剂量、液滴数字PCR等方法,检测孕8周后的胎儿是否携带单基因疾病致病基因的致病性突变[27-28]。目前已有对表现为低位耳、小耳畸形的胎儿进行NIPT检测,发现NS致病基因突变的临床案例[29]。随着检测方法不断优化,临床上通过NIPT进行更多MAS产前基因诊断将成为可能。已有通过基因编辑技术治疗单基因遗传病的基础和临床前研究,这也是MAS极具前景的研究方向。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2020年2期)2021-01-18 02:51:26
中国生殖健康(2018年4期)2018-11-06 07:12:16
小学生导刊(2018年13期)2018-06-29 03:49:00
现代园艺(2017年21期)2018-01-03 06:41:32
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
医学研究杂志(2015年5期)2015-06-10 06:43:26
现代检验医学杂志(2015年5期)2015-02-06 01:42:20
