
Metal-free multicomponent polymerization toward cationic polyamidines
2022-06-20 08:00MengDuMingLiWangzeSongNanZheng
Chinese Chemical Letters 2022年5期
Meng Du, Ming Li, Wangze Song, Nan Zheng
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China
Keywords:Multicomponent polymerization Metal-free Cationic polymer Polyamidine Water-soluble
ABSTRACT Cationic polymers, also known as polycations, are considered to be the most potential non-viral gene carriers due to their unique advantages such as the ability to bind the negative charge of nucleic acid molecules.Multicomponent polymerization (MCP) is a one-step, tandem strategy to construct complex structures based on multicomponent reactions.Herein, we developed a metal-free MCP method based on three monomers of p-dinitrovinylbenzene (p-DNVB), 1,1-dimethylethyl N,N-dibromocarbamate (BocNBr2),and bis-secondary-amines with a ratio of 1:2:1, to access a library of Boc-substituted polyamidines with well-defined structures and suitable molecular weights (Mw ranging from 4400 Da to 11,000 Da) in high yields (up to 85%) under mild conditions.Upon the removal of Boc groups, a series of water-soluble polymers with cationic property were prepared and their gene binding capability was further evaluated.
Multicomponent reaction (MCR), in which three or more starting materials participated into a reaction to afford a complex molecule in a one-pot and time-saving manner, shows great potential and strongly fascinates the chemists [1,2].MCR is a cascade strategy for quickly obtaining versatile and important motifs with highly functional structures [3], such as amides [4,5],propargylamine [6],N-sulfonylamidines [7] and dihydropyrimidin-2H-ones [8].Multicomponent polymerization (MCP), derived from MCR, is a burgeoning area in polymer chemistry with unique advantages including structural diversity and operational simplicity.Compared with the traditional polycondensation methods using two monomers, MCP dramatically broadens the versatility of the polymers through the involvement of different combinations of three or more monomers [9].Till now, numerous types of MCRs, including the Passerini, Ugi, Biginelli and Cu-catalyzed azide-alkyne cycloaddition (CuAAC) reactions have been successfully reassessed and disclosed by polymer chemists to prepare novel and functional polymers [3,7,10-13].However, further development and application of MCPs are always hindered by the limited MCRs suitable for MCPs, narrow scope of monomers, low solubility of the obtained polymers as well as the undesired side reactions caused by the introduction of incompatible monomers.Therefore, it is of great significance to develop new MCP methods to prepare functional polymers with well-defined structures and desired molecular weights (Mws).
Polyamidine is a kind of cationic polymer containing amidine groups as the repeating unit.Amidine, also called iminoamide,is not only a useful structure existing in numerous bioactive molecules, but also widely used in agrochemicals, drugs, as well as the pharmaceutical and fine chemical industries [14-19].Even though various strategies have been reported to prepare amidines,most of the methods were based on toxic reagents such as isonitriles, nitriles, azides, or transition-metals like Pd, all of which were environmental-unfriendly [20-24].Moreover, few of them have been utilized in constructing polyamidine, let alone the efficient preparation of polyamidine in a relatively green manner.Some MCP methods have been reported using Mannich reaction starting from formaldehyde or formalin to prepare cationic polymers, such as polyamines [25,26].As a derivative of aminecontaining polymer like polyvinylamine or polyethyleneimine,polyamidine features the advantages of good thermal stability,low toxicity, and high positive charge density [27].Besides that,polyamidine as a typical cationic polymer, also exhibits the potential as the ideal non-viral gene delivery vector to condense and delivery negatively charged nucleic acids [28,29].
In 2019, Li and An’s groups reported a metal-free MCR starting fromtrans-β-nitrostyrene derivatives, dibromo amides, and various amines to rapidly construct amidine frameworks with high diversity and complexity [30].Inspired by such method, in this work,a metal-free MCP was developed usingp-dinitrovinylbenzene(p-DNVB),N,N-dibromocarbamate (BocNBr2), and bis-secondary amines to access a library oftert–butyloxycarbonyl (Boc)-substituted polyamidines (Scheme 1A).Since Boc could be facilely cleaved by trifluoroacetic acid (TFA), a library of cationic polyamidines with non-fully substituted amidines and watersoluble property could be afforded upon the removal of Boc groups.Such cationic polyamidines could be potentially utilized in the gene delivery area as the non-viral vectors due to the high density of positive charges on the main chain (Scheme 1B).
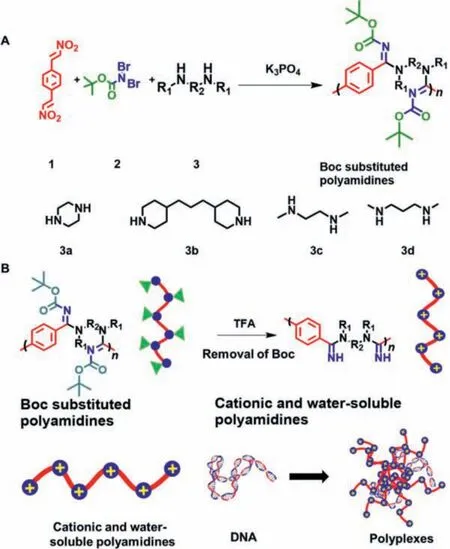
Scheme 1.(A) Synthetic route of the polyamidine library using MCP from p-DNVB,BocNBr2 and bis-secondary-amines.(B) Preparation of cationic polyamidines and formation of polyplexes with plasmid DNA.
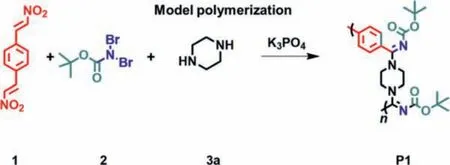
Scheme 2.Model MCP using 1, 2 and 3a as monomers.
Prior to the optimization of polymerization condition,p-DNVB 1 and BocNBr22 were initially synthesized and purified following the methods reported before [31,32] (Schemes S1 and S2, Figs.S1-S4 in Supporting information).The model MCP between 1, 2 and commercially available piperazine 3a was firstly performed to afford the targeting polymer with moderateMwof 3600 g/mol and yield of 92.5% at the optimized conditions reported by Li [30].To further improve the polymer’sMw, effect of temperature (10–60°C), monomer concentration (0.05–0.3 mol/L), polymerization time(12–48 h), and solvent (DMF, MeCN, MeOH, THF, DCE, CHCl3and toluene) were systemically evaluated to optimize the polymerization conditions (Scheme 2).
The effect of temperature was firstly studied by performing the polymerization in CHCl3at the feeding ratio of [1]:[2]:[3a]=1:2:1 under various temperatures.As shown in Table 1, high temperature could notably increase theMwof polymers and promote such MCP.It was mainly because of the more improved reactivity and bettersolubility of the monomers at higher temperature.As a result, 60°C was screened to be the optimal temperature and used in the further experiments.The monomer concentration played a crucial role in most of MCP.As shown in Table 2, theMwof the polymers peaked (5600 g/mol) at the monomer concentration of 0.1 mol/L.Both low monomer concentration (0.05 mol/L) and high monomer concentration (>0.15 mol/L) led to the decrease ofMw.When the monomer concentration was as low as 0.05 mol/L, theMwwas only 4700 g/mol although the yield of the polymer could reach 70%.The possible reason was that diluted concentration would hinder the molecular collision between monomers.When the concentration of the monomer was higher than 0.1 mol/L, the yield andMwwould also decrease, because the high concentration would lead to a fast initial reaction rate to quickly form a large number of oligomers.The solubility of such oligomers would severely decrease at high concentrations to affect the final yield andMw.Therefore, all the following MCP experiments were performed with a concentration of 0.1 mol/L at 60 °C.The polymerization time was subsequently investigated and the results in Table 3 clearly indicated that theMwof the polymer quickly reached 4400 g/mol at the first 12 h and further increased to 5600 g/mol when elongating the polymerization time to 48 h.Considering that continuously extending the reaction time would not remarkably improve theMw, 48 h was screened to be the optimal polymerization time and used in the further experiments.
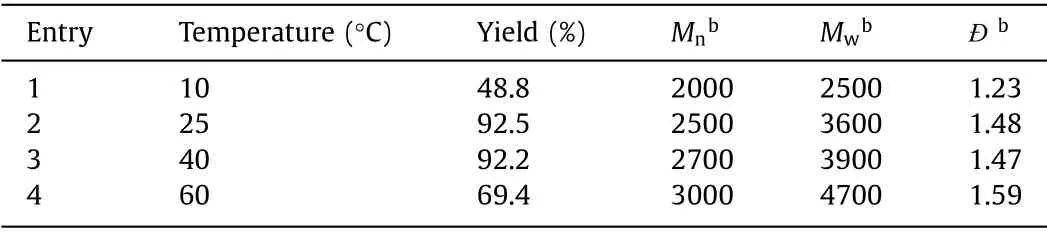
Table 1 Effect of temperature on the MCP of 1, 2 and 3a.a
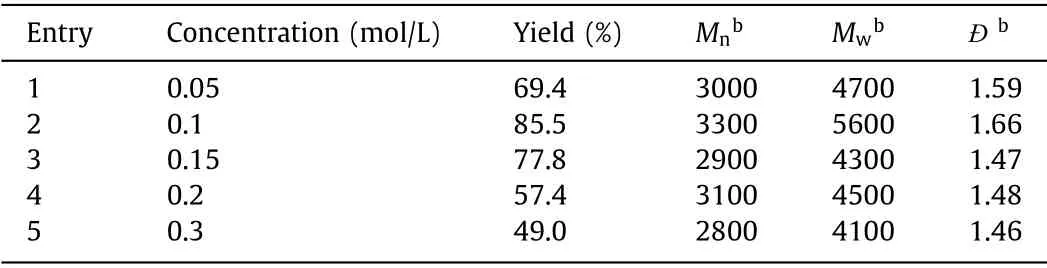
Table 2 Effect of monomer concentration on the MCP of 1, 2 and 3a.a
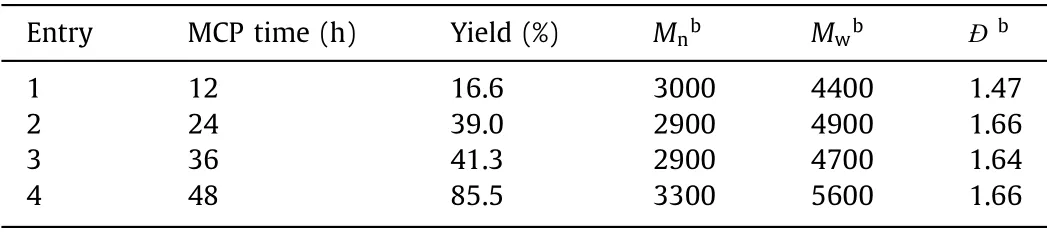
Table 3 Effect of polymerization time on the MCP of 1, 2 and 3a.a
Solvent effect was finally evaluated since the polarity of the solvents would also affect the polymerization results.As shown in Table 4,N,N-dimethylformamide (DMF), dichloroethane(DCE), chloroform (CHCl3), acetonitrile (MeCN), methanol (MeOH),tetrahydrofuran (THF) and toluene with different polarities were selected to optimize the solvent and the results indicated that suchMCP could well tolerate most of the organic solvents and polymers with moderateMw(around 5000 g/mol) could be afforded.CHCl3was screened as the optimal solvent for the further experiments because of the relatively higher yield (85.5%) andMw(5600 g/mol)compared with other solvents.
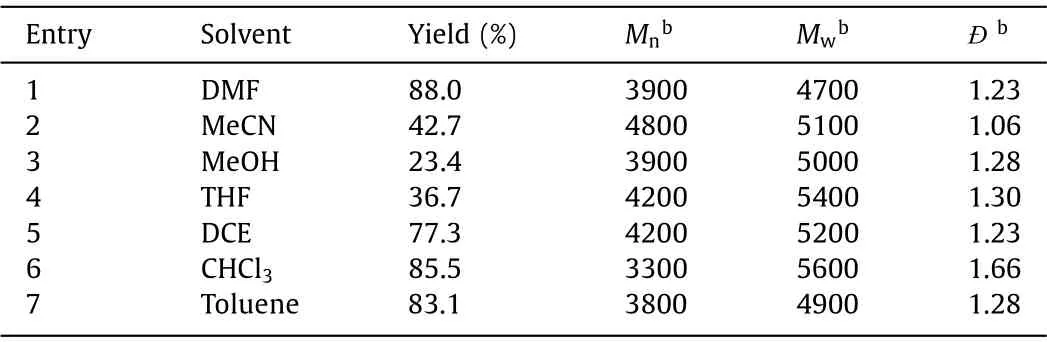
Table 4 Effect of solvent on the MCP of 1, 2 and 3a.a

Table 5 Effect of BocNBr2 concentration on the MCP of 1, 2 and 3a.a
Finally, the concentration of BocNBr2was optimized to evaluate whether increasing the amount of BocNBr2would further improve the Mw.Unfortunately, the results in Table 5 indicated that the optimized ratio ofp-DNVB top-DNVB was 2.Further increasing the amount ofp-DNVB would lead to severe solubility issue and decrease theMw.
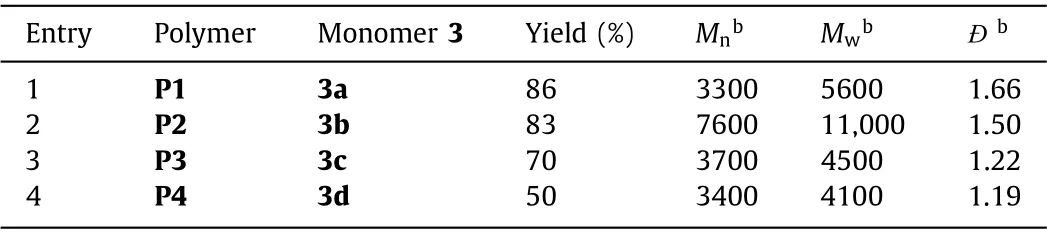
Table 6 MCP results of p-DNVB, BocNBr2 and various bis-secondary-amines.a
To gain exact structure information of the products, both the1H NMR and13C NMR spectra of P1 and the corresponding monomers 1, 2, 3a were shown in Fig.1 as a model example.Comparing the1H NMR spectra of P1 and 1 (Fig.1A), the peaks of -CH=CH- for nitroalkene atδ8.17 andδ8.35 corresponding to 1 completely vanished in the1H NMR spectrum of P1, demonstrating the completed consumption of the 1 monomer and the high conversion rate in MCP.The peaks atδ1.50 associated with thetert–butyl protons of 2 notably appeared in P1, indicating that 2 successfully participated into the MCP.The peak atδ2.77 representing the protons adjacent to the secondary amine in 3a slightly moved to low fields, indicating that the electron-withdrawing ability of the amidine group was stronger than the secondary amine, which reduced the electron cloud density around the methylene group, causing the chemical shift of the methylene hydrogen to move to low field.Comparing the13C NMR spectra of P1 and 1 (Fig.1B), the peak of -CH=CH- atδ137.9 andδ139.3 corresponding to 1 completely vanished in the13C NMR spectrum of P1, which was in consistent with the1H NMR result.The peaks atδ156.6 associated with the-C=O- ontert–butyl of 2 appeared on the spectrum of P1 at the same position without obvious shifts, indicating that 2 participated into the MCP and the final polymer had the Boc functional group.In the same way, the peaks atδ47.0 associated with the -CH2-of 3a also appeared on the spectrum of P1 at the similar position.More importantly, the typical carbon peak of -C=N on the amidine group emerged at aroundδ161.8, demonstrating that the MCP was successful.1H NMR spectra and13C NMR spectra data proved that all the three components, 1, 2, and 3a totally participated into the MCP to form the targeting (Boc)-substituted polyamidine.In order to further confirmed the structure, FT-IR spectra of 1, 2, 3a and polymer P1 were performed (Fig.1C).Comparing the spectra of P1 and 1, the peaks of =C-H at 3100 cm-1, -C=C- at 1633 cm-1, -NO2at 1500 cm-1and 1335 cm-1corresponding to 1 disappeared in the FT-IR spectrum of P1.In addition, the peaks of Ar-H at 3040 cm-1and 720 cm-1were obviously found in P1, demonstrating 1 participated in MCP.The peaks of -COO- at 1700 cm-1,-C(CH3)3at 1395 cm-1and 1365 cm-1corresponding to 2 perfectly existed in the FT-IR spectrum of P1, showing that 2 participated in MCP and the final product containing the Boc group.Comparing the spectra of P1 and 3a, the peak of -N-H at 3210 cm-1was vanished while the peaks of -CH2- at 2930 cm-1and -C-N- at 1150 cm-1still existed in the polymer, showing that 3a was also involved in the polymerization reaction.
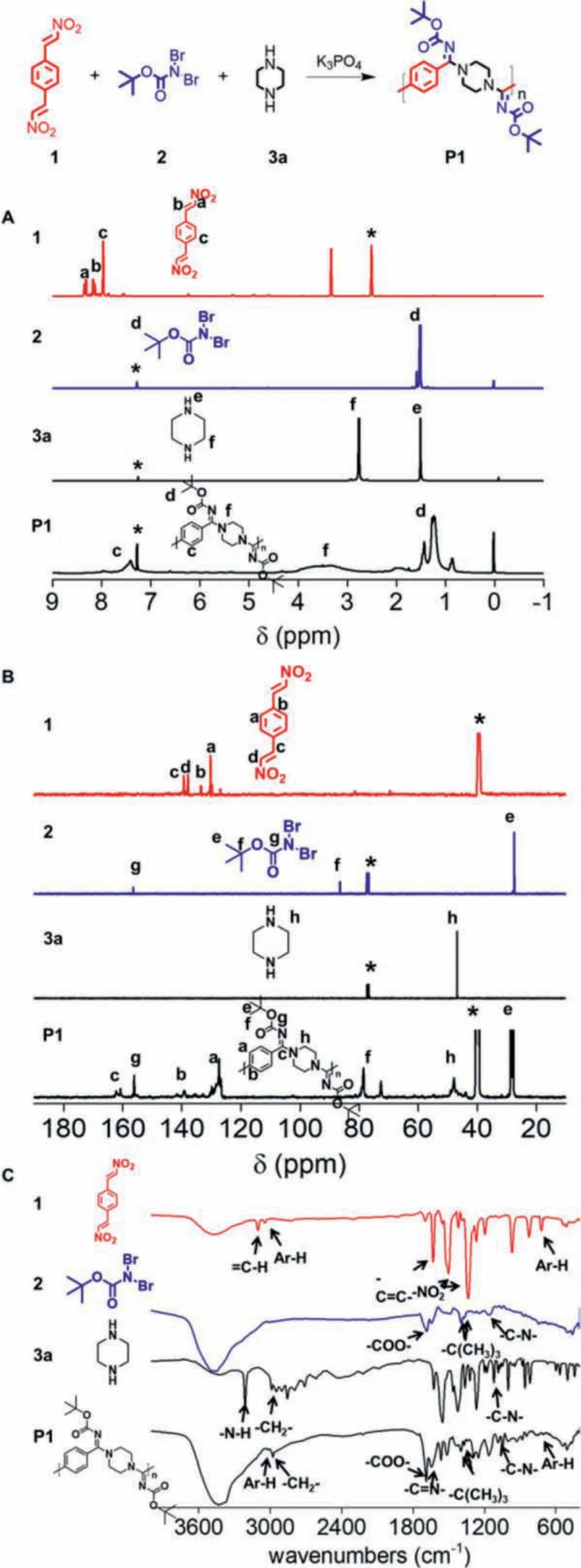
Fig.1.Structural characterization of model MCP product P1 using 1, 2, and 3a as monomers.(A) 1H NMR spectra of 1, 2, 3a and polymer P1 in DMSO–d6 (1) and CDCl3 (2, 3a, P1).The peaks of related solvents are marked with asterisks.(B) 13C NMR spectra of 1, 2, 3a, and polymer P1 in DMSO–d6 (1, P1) and CDCl3 (2, 3a).The peaks of related solvents are marked with asterisks.(C) FT-IR spectra of monomer 1, 2, 3a and P1.
With the optimized conditions at hand, four kinds of commercially available bis-secondary amines with different steric hindrance, 1,4-diazacyclohexane (3a), 1,3-di-4-piperidylpropane(3b),N,N-dimethyl-1,2-ethanediamine (3c) andN,N-dimethyl-1,3-propanediamine (3d) were explored to further expand the substrate scope and demonstrate the robustness of such MCP (Scheme 1A).A library of Boc-substituted polyamidines with similar pendants but various backbone structures were facilely prepared and fully characterized (Figs.S5-S9 in Supporting information, Table 5).As shown in Table 6, we found that cyclic bis-secondary amines(3a and 3b) could promote such MCP to afford the polymer with relatively high yields (>80%) andMw(>5000 g/mol) compared to linear monomers.3b with less steric hinderance effect exhibited highestMw(11,000 g/mol), meaning that cyclic secondary amines with flexible chains favored such amidination polymerization.
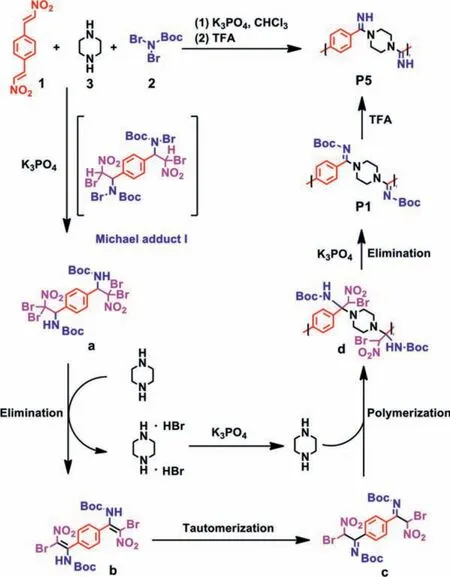
Scheme 3.Mechanism of MCP to afford cationic polyamidine.
To explore whether the (Boc)-substituted polyamidine could be furthermore modified to prepare the cationic and water-soluble polyamidine, TFA was used to remove Boc in P1 and form the non-fully substituted polyamidine, P5 (Fig.2).After P1 was thoroughly deprotected by TFA, P5 exhibited excellent water solubility at the pH values lower than 7.4.The chemical structure of P5 was evaluated using1H NMR and FT-IR spectra.As shown in Fig.2A, the peaks at the range ofδ1.2 associated with thetert–butyl protons of P1 totally disappeared, which verified the successful deprotection of Boc group from P1.In addition, as shown in Fig.2B,the peaks of -COO- at 1700 cm-1and -C(CH3)3at 1395 cm-1and 1365 cm-1corresponding to Boc group in P1 vanished in P5, and the broad peak at 3400 cm-1indicating -N-H appeared.All the data showed that a kind of water-soluble and cationic polyamidine could be successfully prepared using such MCP method, followed by a step of Boc removal.
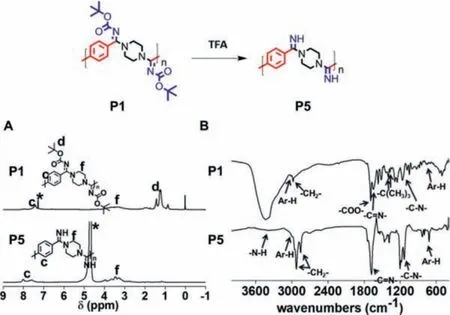
Fig.2.Preparation of cationic polyamidine upon the removal of Boc group.(A) 1H NMR spectra of P1 and P5 in CDCl3 (P1) and D2O (P5).The peaks of related solvents are marked with asterisks.(B) FR-IR spectra of P1 and P5.
Based on the mechanism proposed by Li and An’s groups [30], a possible mechanism for the MCP to prepare the cationic polyamidine was shown in Scheme 3.Firstly, N-Br bond in BocNBr22 was dissociated and adduct I was afforded by Michael addition.I was then transformed into a, followed by an elimination to afford intermediate b bearing bis-enamine, which could be tautomerized to form bis-imine c.Piperazine 3 with bis-secondary amines underwent the nucleophilic addition to bis-imine c, giving the intermediate d, followed by the elimination to provide the polymer P1.Finally, the cationic polyamidine P5 was obtained by the removal of Boc groups using TFA.
Owning to the universal of such MCP and the successful modification mentioned above, four cationic non-fully substituted polyamidine, P5-P8, were subsequently prepared and all the chemical structures were characterized by1H NMR in D2O (Fig.3A and Figs.S10-S13 in Supporting information).Taking advantages of the water-soluble and cationic properties of the obtained polymers,their potentials in gene condensation were evaluated using gel retardation assay.As shown in Fig.3B, only P7 could successfully bind plasmid DNA while P5, P6 and P8 could not facilitate gene condensation.That was mainly because that P7 had the less hindrance around the positive charges of amidine groups compared to P5 and P6 using cyclic amine monomers, which would make the interaction and condensationviaelectrostatic effect more favorable.It was interesting that P7 exhibited excellent DNA binding ability while P8 with similar structures failed, which was possibly due to the odd-even effect [33].Nanocomplex formed by P7 and plasmid DNA at the weight ratio of 20 was further evaluated by DLS, and the result indicated that such complex had the particle size below 200 nm, which was suitable for the further gene delivery application (Fig.3C).Zeta potentials of P7 and the complexes formed by P7 and plasmid DNA were also evaluated by DLS as shown in Fig.3D.It was clearly to find that all the complexes exhibited a positive surface charge when the weight ratios of P7/DNA higher than 5.The zeta potential was around ~30 mV at the weight ratios higher than 20, indicating the successful condensation for the potential gene delivery application.
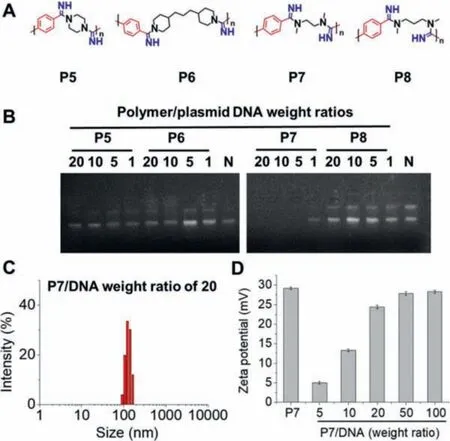
Fig.3.(A) Structures of cationic polyamidines (P5-P8).(B) Gene binding ability of the cationic polyamidines evaluated by gel retardation assay.N represented naked plasmid DNA.(C) DLS results of the P7/DNA complexes at the weight ratio of 20.(D) Zeta potentials of P7 and P7/DNA complexes at various weight ratios.
In summary, a mild and metal-free MCP method was developed to prepare a library of Boc-substituted polyamidines using bis-secondary-amine,p-DNVB and BocNBr2.Upon the removal of Boc using TFA and the formation of non-fully substituted amidine groups, cationic polyamidines could be facilely obtained with water-soluble property.Potential gene binding capability was evaluated to afford the optimized structure P7.Further application of such polymer in gene delivery area and the relationship between the structure and gene delivery capability were undergoing in the lab.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Science Foundation of China (No.21978039), Special Funds of the Central Government Leading Local Government for the Technology Development (Nos.2021JH6/10500148, 2021JH6/10500146); Fundamental Research Funds for the Central Universities (Nos.DUT21YG133,DUT20YG120).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.09.031.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction