
Structure-based optimizations of a necroptosis inhibitor (SZM594) as novel protective agents of acute lung injury
2022-06-20 08:00LijunXuYeTuJioLiWnninZhngZhibinWngChunlinZhungLeiXue
Chinese Chemical Letters 2022年5期
Lijun Xu, Ye Tu, Jio Li, Wnnin Zhng, Zhibin Wng, Chunlin Zhung,e,*,Lei Xue
a School of Pharmacy, Second Military Medical University, Shanghai 200433, China
b Department of Medicine, Shanghai East Hospital, Tongji University, Shanghai 200120, China
c Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai 200072, China
d Shanghai Changzheng Hospital, Second Military Medical University, Shanghai 200433, China
e Department of Chemistry, Fudan University, Shanghai 200433, China
Keywords:Acute lung injury RIPK1 RIPK3 Necroptosis Design
ABSTRACT Targeting RIPK1 is a promising strategy for the treatment or alleviation of acute lung injury (ALI).SZM594, a benzothiazole compound previously developed by our research group, possessed good dualtargeting receptor-interacting protein kinase 1 (RIPK1) and RIPK3 activity and anti-necroptosis activity as well as acceptable in vivo efficacy.In this study, the cyclopropyl moiety of SZM594 was modified based on a structure-based design strategy.The resulting cyclohexanone-containing analogue 41 improved the selectivity toward RIPK1 over RIPK3 and the anti-necroptosis activity was also increased compared with those of SZM594.More importantly, compound 41 could inhibit the tumor necrosis factor-α (TNF-α) expression in lipopolysaccharide (LPS)-induced peritoneal macrophage cell model, and significantly alleviate LPS-induced ALI in a mouse model.This compound could significantly inhibit the expressions of the phosphorylation of RIPK1 and down-stream RIPK3 and mixed lineage kinase domain-like protein (MLKL).Thus, these cyclohexanone-containing benzothiazole analogues represent promising lead structures for the discovery of novel protective agents of ALI.
Acute lung injury (ALI) is an acute and life-threatening inflammatory disease associated with high morbidity and mortality worldwide [1].ALI is caused by multiple diseases in clinic,such as sepsis, pneumonia, major trauma, and severe acute respiratory syndrome coronavirus (SARS-CoV) and SARS-CoV-2 infectioninduced damages, leading to cell death, short-term dyspnea to respiratory failure and ultimately to acute respiratory distress syndrome (ARDS) [2–4].However, no effective therapeutic strategies have been developed to mitigate the proinflammatory response in ALI [1,5].Thus, there is an urgent need for the development of drugs for treatment or alleviation of the ALI intercepting the occurrence of the ultimate ARDS [6].
Programmed cell death (PCD), including necroptosis, is increasingly recognized as a critical driver in the development of ALI [7–9].Receptor-interacting protein kinase 1 (RIPK1) has been reported to regulate necroptosis, in response to a broad set of inflammatory stimuli in human diseases [10–14].With the first disclose of necrostatin–1 (Nec–1) by Yuan group, RIPK1 has been confirmed as a prominence among therapeutic targets [15,16].RIPK1 is activated in response to various cell signals, including the activation of the death receptors TNF receptor 1 (TNFR1), Fas ligation (FasL), and TNF-related apoptosis-inducing ligand (TRAIL) [17,18].For instance,in the tumor necrosis factor-α(TNF-α) stimulated cells, RIPK1 activation executes the process of apoptosis and necroptosis [19].Inhibition of caspase-8 activity by Z-Val-Ala-Asp-(OMe)-fluoromethyl ketone (z-VAD-FMK) allows recruitment of RIPK3, interacting with RIPK1 to form a necrosome.The necrosome recruits and phosphorylates mixed lineage kinase domain-like protein (MLKL), leading to its oligomerization and plasma membrane localization, resulting in rupture of the cell membrane, lytic cell death with subsequent release of damage associated molecular patterns (DAMPs),which can cause various inflammatory diseases [20–22].Therefore,blocking RIPK1 activation has been a promising strategy to improve the outcome of ALI [20].Recent studies demonstrated that Nec–1 could suppress lipopolysaccharide (LPS)-induced ALI and ischemia/reperfusion (I/R)-induced lung injury in animal models(Fig.1A) [23,24].Nec-1 was also confirmed to markedly reduce neutrophil-dependent inflammation in LPS-induced ALI mice [25].Compound Nec-5 (a RIPK1 inhibitor) also reduced the severity ofS.marcescenspneumonia in a mouse intratracheal challenge model[26].However, these compounds are only applied as tool compounds and the poor pharmacokinetic properties (t1/2<5 min in a mouse microsomal assay) and narrow structure–activity relationship (SAR) profile limit their clinic applications [27].
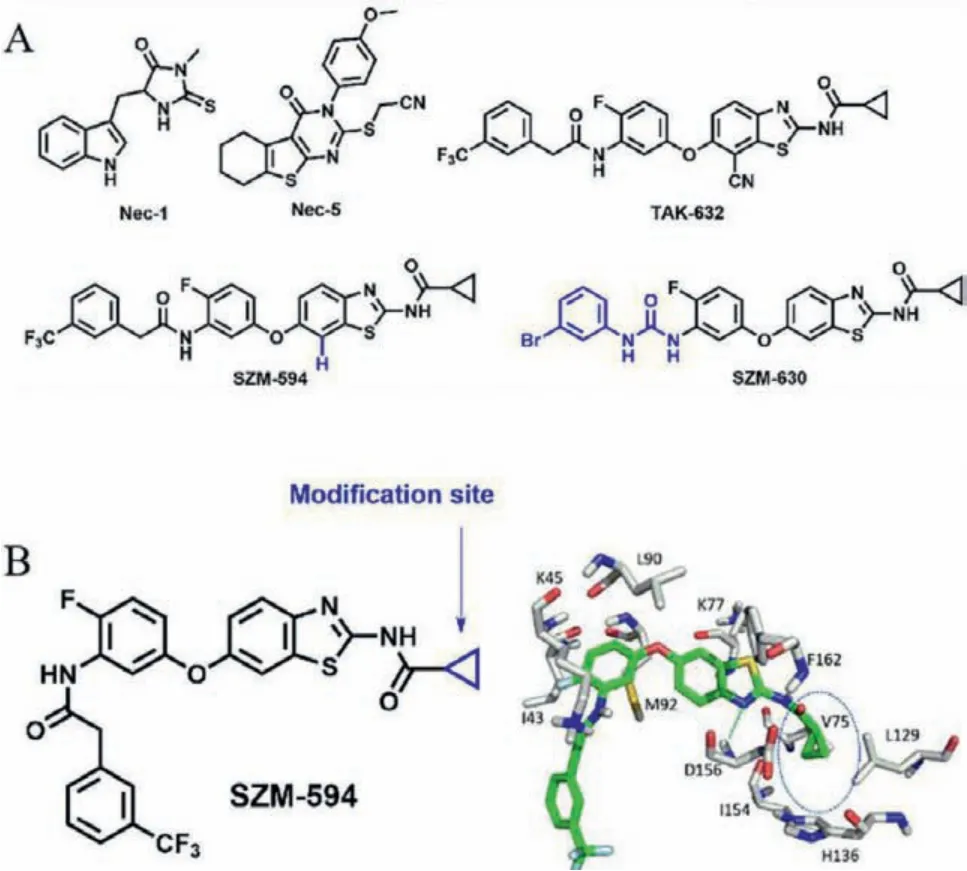
Fig.1.(A) Chemical structures of representative necroptosis inhibitors.(B) The predicted binding mode of SZM-594 with RIPK1 and the proposed modification site(highlighted in blue).
TAK632, a benzothiazole compound, was screened by our group from an in-house fluorine-containing compound library by targeting RIPK1 and RIPK3 [28].With the ligand-based structure optimization, we obtained a cyano-free compound SZM594 showing improved RIPK1/RIPK3 inhibitory activity and a selective RIPK3 inhibitor SZM630 was also obtained by modifying the benzyl part[28,29].Thus, in the present study, we would like to develop the SZM594 analogues by the structure-based optimization with the purpose to improve the selectivity toward RIPK1 over RIPK3 and inhibitory activity toward cell necroptosis.Finally, we obtained compound 41 with better anti-necroptotic activity (3.4-fold) and selectivity (35-fold) than the lead compound SZM594.It inhibited the TNF-αexpression in LPS-induced peritoneal macrophage cell model, and significantly reduced LPS-induced inflammation and alleviated ALI in a mouse model.This compound could significantly inhibit the phosphorylations of RIPK1, RIPK3 and MLKL.
On the basis of our previous docking result (Fig.1B) [28,29], a key hydrogen-bonding interaction between the benzothiazole ring and protein residue D156 and strongπ-πstacking interactions with F162 were predicted which was considered to be kept in the structure optimizations.The aniline group was inserted into the kinase gatekeeper region (I43, K45 and M92 residues), making the trifluorophenyl group point to the solvent exposed region.The cyclopropyl group was located in a hydrophobic cavity (V75 and L129 residues).Thus, these two terminal sites of SZM594 should be modified.Previously, the trifluorophenyl terminal was replaced by various carbamides to obtain highly potent RIPK3 selective inhibitors [29].In the present study, we focused on the cyclopropyl group for optimization with a optimization strategy.
The first RIPK1 inhibitor Nec-1 was identified using a phenotypic cell screening that measured the ability to block necroptotic death [15,30].Thus, in this study, we applied a human colon adenocarcinoma cell (HT-29) model co-treated by a combination of TNF-α, second mitochondrial-derived activator of caspases(Smac) mimetics, and z-VAD-FMK to form the necroptosis model,termed as TSZ-induced HT-29 cell model.The inhibitory effects of the compounds on RIPK1 and RIPK3 kinases were detected by a KINOMEscanTMassay [28].
First, the cyclopropyl group was replaced by simple alkyl groups(1-6, Table S1 in Supporting information).Compound 1 with a methyl group showed decreased anti-necroptosis activity and RIPK1/3 binding activity compared with those of SZM594.The activity was slightly improved with one more carbon chain (ethyl,compound 2).With the increase of the steric hindrance, the activity toward necroptosis and RIP kinases were gradually decreased(compounds 3-6).Compound 7 with theα-hydrogens replaced by fluorine atoms totally lost the anti-necroptosis activity at 10 μmol/L.Besides, the carbonyl (8), sulfonyl (9) and hydroxyl (10)substitutions were also introduced into the alkyl group.Compounds 8 and 10 exhibited comparable anti-necroptosis activities with SZM594 and compound 9 was much less active, suggesting that the hydrogen donor/acceptors might exist around the hydrophobic cavity and probable carbon chains were required.Apart from these alkyl substitutions, a series of cycloalkyl substituted compounds also were synthesized (compounds 11-14).Their antinecroptosis activities were gradually decreased with the increase of the ring members [cyclobutyl (11)>cyclopentyl (12)>cyclohexyl (13)>cycloheptyl (14)].Consistently, the inhibitory activity toward RIPK1 and RIPK3 followed the same sequence showing, to some extent, the RIPK1 selectivity.
Next, we continued to synthesize a series of substituted cycloalkyl and heterocyclic analogues (Supporting information Table S2).Compound 15 with a 2-methylcyclopropyl group maintained anti-necroptosis and RIPK inhibitory activity compared with those of SZM594.Compound 16 with α-fluorine atoms showed a decreased 50% effect concentration (EC50) value of 1120 ±410 nmol/L.Compound 17 with a 2-fluorocyclopropyl had an EC50value of 170 ± 3 nmol/L.The anti-necroptosis activity and inhibitory activity toward RIPK1 and RIPK3 of compound 18 were decreased when one more fluorine atom introduced.Then,compounds with substituted four-membered rings were synthesized.Compound 19 with 3,3-dimethylcyclobutyl, 20 with 3,3-difluorocyclobutyl, and 21 with Boc-azetidine-3-yl exhibited similar cell- and kinase-based activity.Differently, the kinase inhibitory activity was less active toward RIPK3 than RIPK1.Compound 22 with a tetrahydrofuran-3-yl group showed an EC50of 280 nmol/L and aKdvalue of 220 nmol/L toward RIPK1 and 1800 nmol/L toward RIPK3.Like compound 16, a tetrahydrofuran-2-yl substitution made compound 23 significantly decreased the anti-necroptosis activity (EC50= 2940 ± 580 nmol/L).Compounds with furan (24)and thiophene (25) showed the activity of about 500 nmol/L.For compounds 26–33, the cell-based activities were decreased along with the enlargement of the rings and substitutions, except compound 28 (EC50= 190 ± 30 nmol/L).For the RIPK1/3 binding activity, these compounds showed good activity toward RIPK1 and no apparent activity was observed toward RIPK3 at 5000 nmol/L,providing important SAR information for the next design.
With the information of Tables S1 and S2, the introduction of carbonyl or hydroxyl groups into appropriate alkyl groups might benefit for the activity.Therefore, compounds 34–42 with carbonyl or hydroxyl substitutions on the cyclobutyl, cyclopentyl or cyclohexyl groups were synthesized (Table 1).As a result, compounds with carbonyl showed much better anti-necroptotic activity than their corresponding analogues with 3-hydroxyl groups.Compound 34 with 3-oxocyclobutyl had the best anti-necroptosis activity with an EC50of 40 ± 6 nmol/L, however, the binding activity toward RIPK1/3 was decreased compared with SZM594.Compounds 36 and 37 with cyclopentyl rings showed better activity toward RIPK1 but much lower activity toward RIPK3 (8–12-fold).When introducing substitutions on theα-positions of the pentyl rings (38, 39),the activity was decreased or totally lost.Similar as previous analogues, compound 40 with larger acetyl group on the cyclopentyl ring reduced the activity with an EC50value of 1430 ± 90 nmol/L.Compound 41 with a cyclohexanonyl group had an EC50of 50 ±2 nmol/L and the hydroxylhexanonyl compound 42 had an EC50of 120 ± 9 nmol/L.They had a higher RIPK1 activity (Kd= 83 and 77 nmol/L) and the activity toward RIPK3 was much lower(Kd= 2900 and 3000 nmol/L).Last mention, all these compounds did not show apparent cytotoxicity at 50 μmol/L that was an improvement compared with SZM594 [50% cytotoxic concentration(CC50) = 36.5 μmol/L].
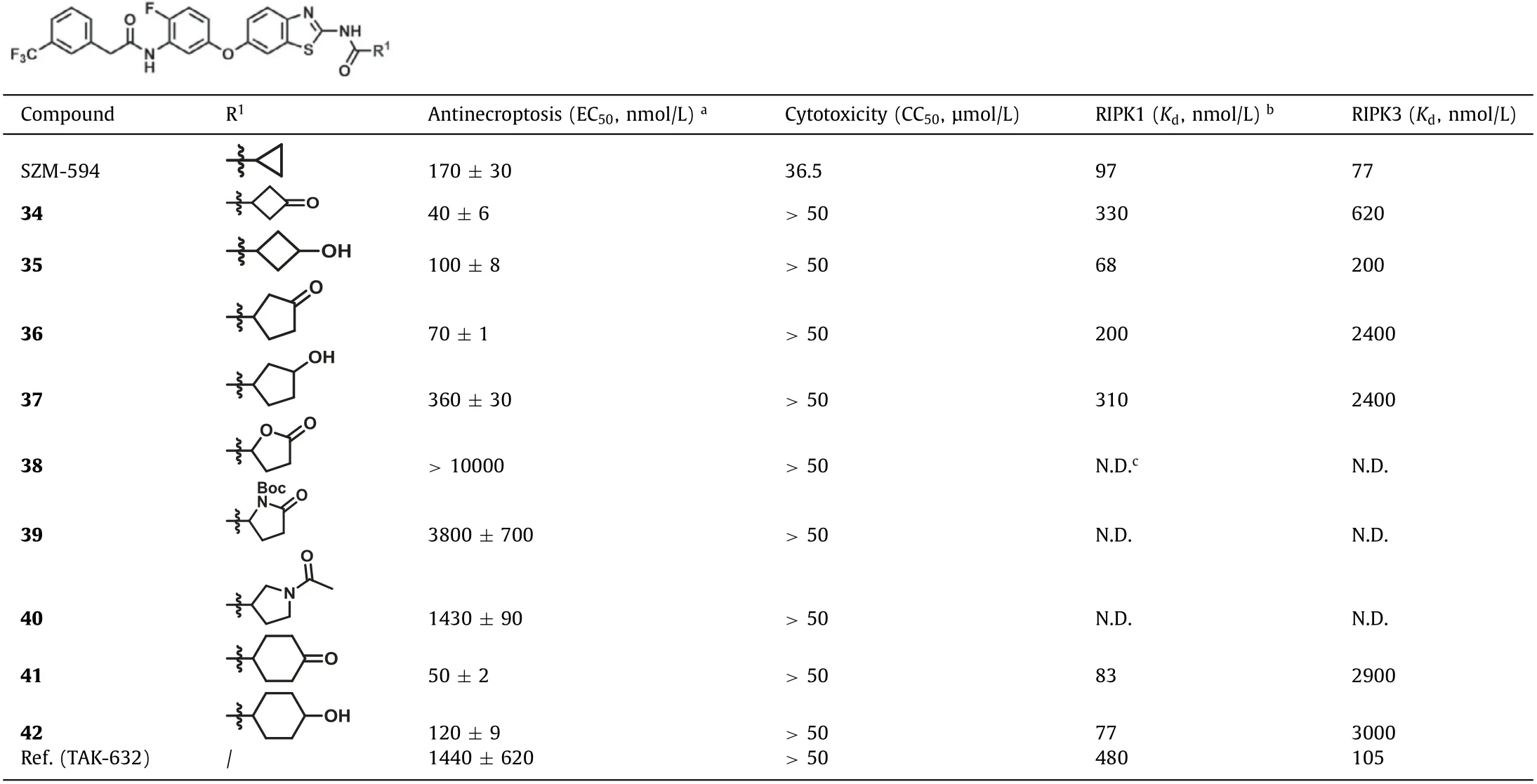
Table 1 The third round of optimizations.
Considering the inhibition of necroptosis could be a promising strategy for the treatment of inflammatory diseases [31–34], the compounds (34, 36 and 41) in Table 1 with potent anti-necroptosis activity (EC50<100 nmol/L) were screened for their ability to inhibit TNF-αthat induced by LPS in mouse peritoneal macrophages.They were evaluated to inhibit the elevation of TNF-αat 1 μmol/L showing more than 50% inhibition for the next step.Furthermore,we found that the three compounds dose-dependently (1, 5 and 10 μmol/L) inhibited TNF-αrelease (Fig.S1 in Supporting information).Compound 41 showed the best TNF-αinhibition by ~80% at 10 μmol/L, which was better than 34 and 36.
Considering the satisfactory activity of compound 41 including anti-necroptosis (EC50= 50 ± 2 nmol/L), RIP kinase (Kd,RIPK1= 83 nmol/L,Kd,RIPK3= 2900 nmol/L) and selectivity (35-fold), and TNFαinhibition (~80% at 10 μmol/L), we then further evaluated its antinecroptotic specificity in cells.It dose-dependently restored cell viability from TSZ-induced necroptosis in human HT-29 cells(Fig.2A).It also protected against TNF-α, cycloheximide, and z-VAD-fmk (TCZ)-induced necroptosis (Fig.2B).However, it did not protect cells from TNF-αand cycloheximide (TC)- or TNF-αand Smac mimetic (TS)-induced apoptosis (Figs.2C and D).In murine L929 cells, we also found that compound 41 protected against necroptosis induced by TNF-αand z-VAD-fmk (TZ) in a dosedependent manner (Fig.2E).The above results indicate that compound 41 inhibits the necroptosis rather than apoptosis pathway.
To investigate the inhibitory mechanism of compound 41, we examined phosphorylation of RIPK1, RIPK3 and MLKL in TSZtreated HT-29 cells with or without compound 41.Consistent with thein vitrokinase assay, compound 41 completely inhibited the phosphorylation of RIPK1 at 1 μmol/L, resulting in the inhibition of the downstream phosphorylation of RIPK3 and MLKL(Fig.2F).Next, the dose-response manner of compound 41 on the RIPK1/RIPK3/MLKL pathway was also evaluated.Compound 41 inhibited the phosphorylation of RIPK1/3 and MLKL at 4 h in a doseresponse manner (0.1, 0.5 and 1 μmol/L, Fig.2G).Since the phosphorylation of RIPK1 and RIPK3 is required for RIPK1–RIPK3 necrosome formation [35], we then explored the formation of necrosome in HT-29 cells after pretreating with compound 41, and we found that compound 41 completely blocked TSZ-induced necrosome formation at 6 h (Fig.2H).For the immunoprecipitation, a similar phenomenon was observed (Fig.2I).Thus, compound 41 effectively blocks necrosome formation by inhibiting TSZ-induced phosphorylation of RIPK1/RIPK3/MLKL pathway.
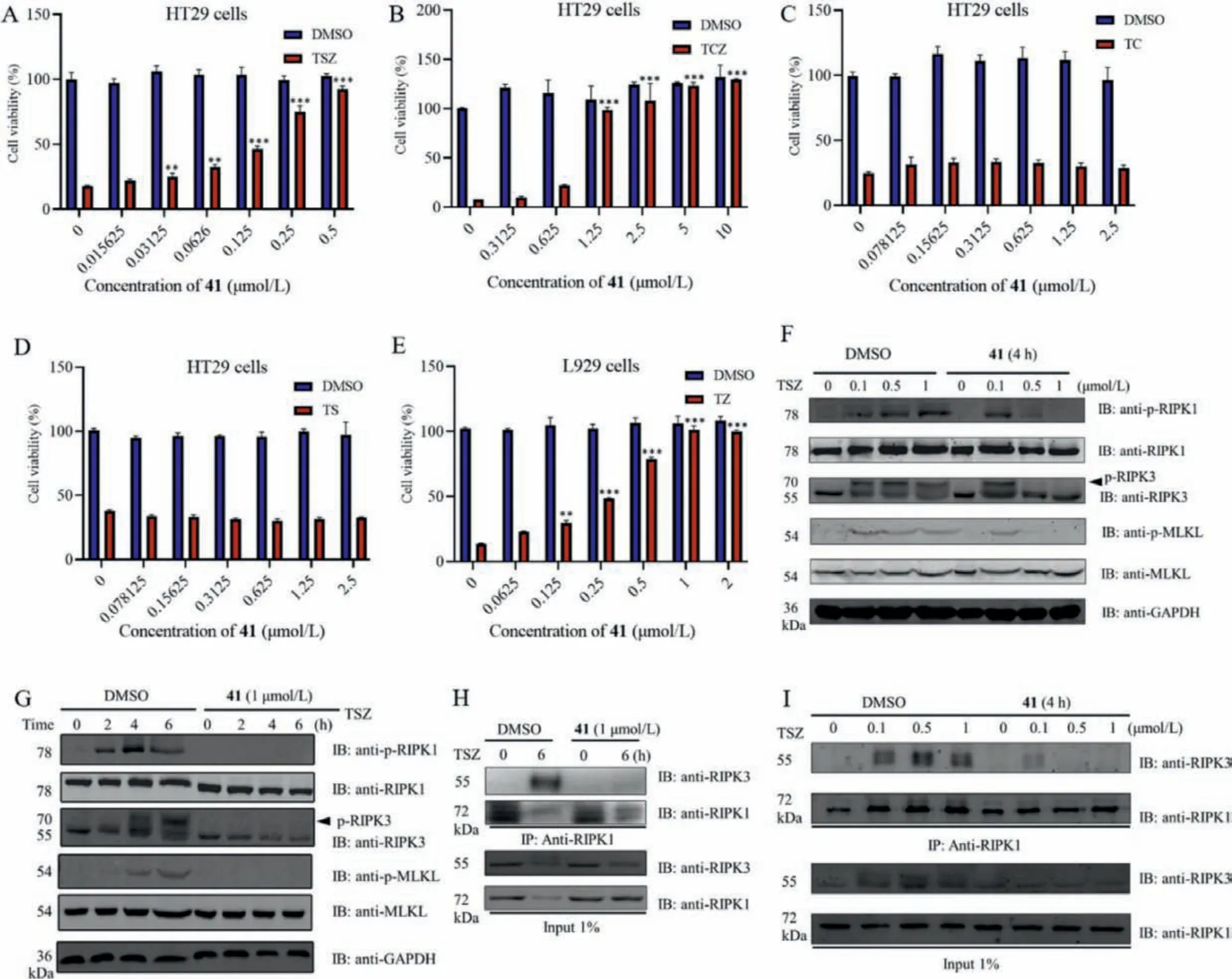
Fig.2.Anti-necroptosis effects of compound 41 in vitro.(A) HT-29 cells were treated with compound 41 as indicated concentrations followed by stimulation with TNF-α (20 ng/mL), Smac (10 nmol/L) and z-VAD-fmk (20 μmol/L) (TSZ) for 12 h.(B) HT-29 cells were treated with various concentrations of compound 41 as indicated by stimulation with TNF-α (20 ng/mL), cycloheximide (5 μg/mL) and z-VAD-fmk (20 μmol/L) (TCZ) for 16 h.(C) HT-29 cells were treated with various concentrations of compound 41 as indicated by stimulation with TNF-α (20 ng/mL) and cycloheximide (5 μg/mL) (TC) for 24 h.(D) HT-29 cells were treated with compound 41 as indicated concentrations followed by stimulation with TNF-α (20 ng/mL) and Smac (10 nmol/L) (TS) for 30 h.(E) L929 cells were pretreated with compound 41 as indicated concentrations followed by stimulation with TZ for 12 h.Results shown are means ± SD from three independent experiments.*P <0.05, **P <0.01, ***P <0.001 versus TSZ, TCZ or TZ simulation without compound 41 treatment.(F) HT-29 cells were pretreated with compound 41 (1 μmol/L) and then stimulated with TSZ at the indicated time points.Cells were lysed and immunoblotted with the indicated antibodies.(G) HT-29 cells were pretreated with compound 41 (0, 0.1, 0.5 and 1 μmol/L) and then stimulated with TSZ for 4 h.Cells were lysed and immunoblotted with the indicated antibodies.(H) HT-29 cells were treated with DMSO or compound 41 (1 μmol/L) under the stimulation of TSZ for 6 h.(I)HT-29 cells were treated with DMSO or compound 41 with indicated concentrations under the stimulation of TSZ for 4 h.The cell lysates were immunoprecipitated with an anti-RIPK1 antibody (IP:RIPK1) and analyzed by immunoblotting with the indicated antibodies.GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
To explore whether compound 41 protects against RIPK-driven inflammationin vivo, we tested this compound in a TNF-αcombined with the caspase inhibitor zVAD co-induced systemic inflammatory response syndrome (SIRS) model, leading to an earlier onset systemic inflammatory response in about 3 h mainly characterized by hypothermia [36].The efficacy of compound 41 could be measured by the ability to prevent mouse body temperature loss.The compound was injected intraperitoneally (i.p.) 1 h before intravenous (i.v.) injection of mouse tumor necrosis factor-α(mTNFα) and i.p.injection of z-VAD, and showed 65.5% and 67.7% protection from temperature loss over 3 h, at doses of 5 and 10 mg/kg,respectively (Fig.3A).
To investigate the role of compound 41 in ALI, an LPS-induced mouse model of ALI was established following our previous study[3].Compound 41 (10 and 20 mg/kg) was injected (i.p.) 0.5 h before the injection of LPS (i.p., 15 mg/kg).Considering that the cytokine level in bronchoalveolar lavage fluid (BALF) can directly reflect inflammatory conditions of the lung [37], the pulmonary inflammatory cytokine levels were detected by ELISA.As shown in Fig.3B, the levels of TNF-αand interleukin-1β(IL-1β) in BALF in compound 41-treated mice are lower than those of model mice.Meanwhile, serum TNF-αwas also significantly reduced after compound 41 treatment at the high concentration.At the mRNA level,compound 41 also inhibited the expression of inflammatory genes of TNF-α, IL-1βand interleukin-6 (IL-6) in the lung tissue of mice(Fig.3C).These findings indicated that inflammations of ALI were apparently alleviated in the compound 41 treated mice.Consistent with thein vitroRIPK1 inhibitory activity, compound 41 also exhibited to almost completely block the phosphorylation of RIPK1 in the lung tissue of ALI model mice (Fig.3D), indicating that the anti-inflammatory efficacy was produced by the inhibition of RIPK1in vivo.
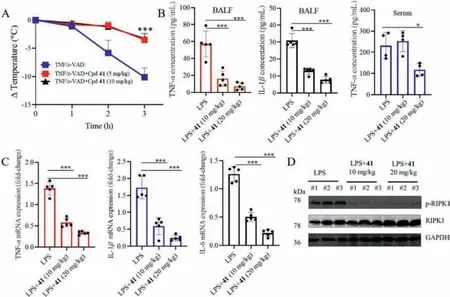
Fig.3.Compound 41 protected mice from TNF-α induced systemic inflammatory response syndrome (SIRS) and LPS-induced ALI in vivo.(A) Body temperature curve of C57BL/6 female mice (n = 10 per group) injected with mTNF-α (100 μg/kg) and z-VAD (1 mg/mL, 250 μL) after pretreatment with different doses of compound 41 (5 and 10 mg/kg).***P <0.001, treatment group vs.vehicle.(B) Levels of secreted inflammatory cytokines isolated from BALF and serum were estimated by enzyme linked immunosorbent assay (ELISA), in order to assess the effects of compound 41 on LPS-induced inflammatory response (n = 5 mice/group).*P <0.05, ***P <0.001 as indicated.(C) mRNA levels of TNF-α, interleukin-1β (IL-1β) and interleukin-6 (IL-6) in lung tissues from LPS-ALI and compound 41 + LPS-ALI mice were detected by reverse transcription-quantitative PCR and were normalized to actin.***P <0.001 as indicated.(D) Suppressive effect of compound 41 on necroptosis pathway in a mouse model of LPS-induced ALI.Western blot of lysates from mice lung tissues was used to estimate the phosphorylation level of necroptosis-associated protein (RIPK1).
In the present study, three rounds of optimizations based on the dual-targeting anti-necroptosis agent SZM594 were performed by a structure-based replacement to obtain the cyclohexanonecontaining analogue 41 with improved the selectivity toward RIPK1 over RIPK3 and the anti-necroptosis activity.This compound could not only inhibit the TNF-αexpression in the LPS-induced peritoneal macrophage cell model, but also significantly reduce LPSinduced inflammation and alleviate ALI, through inhibiting the expression of the phosphorylation of RIPK1, RIPK3 and MLKL.In summary, the cyclohexanone-containing benzothiazole compound represents a promising lead structure for the discovery of novel protective agents of ALI.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was funded by grants from the National Natural Science Foundation of China (Nos.82022065, 81872791, 82073696,81872880 and U20A20136); the Key Research and Development Program of Ningxia (No.2019BFG02017, China); the Scientific and Technological Innovation Project of Science and Technology Commission of Shanghai Municipality (No.21S11900800, China).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.09.059.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction