
A self-assembling prodrug nanosystem to enhance metabolic stability and anticancer activity of gemcitabine
2022-06-20 07:59MiCongGunglingXuShoyouYngJingZhngWnzhngZhngDinshDhumlErikLuriniKiyuZhngYiXiSbrinPriclLingPngWiongZho
Chinese Chemical Letters 2022年5期
Mi Cong, Gungling Xu, Shoyou Yng, Jing Zhng, Wnzhng Zhng,Dinsh Dhuml, Erik Lurini, Kiyu Zhng, Yi Xi, Sbrin Pricl,g, Ling Png,*,Wiong Zho,**
a School of Pharmacy, Xinxiang Medical University, Xinxiang 453003, China
b Henan Key Laboratory of Immunology and Targeted Drugs, School of Laboratory Medicine, Xinxiang Medical University, Xinxiang 453003, China
c Henan Collaborative Innovation Center of Molecular Diagnosis and Laboratory Medicine, School of Laboratory Medicine, Xinxiang Medical University,Xinxiang 453003, China
d Aix-Marseille Université, CNRS, Center Interdisciplinaire de Nanoscience de Marseille, UMR 7325, Equipe Labellisé par La Ligue, Marseille 13288, France
e Chongqing Key Laboratory of Natural Product Synthesis and Drug Research, School of Pharmaceutical Sciences, Chongqing University, Chongqing 401331,China
f Molecular Biology and Nanotechnology Laboratory, Department of Engineering and Architecture, University of Trieste, Trieste 34127, Italy
g Department of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, Lodz 90-236, Poland
Keywords:Self-assembling prodrug Gemcitabine Amphiphilic dendrimer Anticancer candidate Nanomicelles
ABSTRACT Self-assembly is a powerful approach in molecular engineering for biomedical applications, in particular for creating self-assembling prodrugs.Here, we report a self-assembling prodrug of the anticancer drug gemcitabine (Gem) based on amphiphilic dendrimer approach.The prodrug reported in this study demonstrates high drug loading (40%) and robust ability to self-assemble into small nanomicelles, which increase the metabolic stability of Gem and enable entry into cells via endocytosis, hence bypassing transport-mediated uptake.In addition, this prodrug nanosystem exhibited an effective pH- and enzymeresponsive release of Gem, resulting in enhanced anticancer activity and reduced toxicity.Harboring advantageous features of both prodrug- and nanotechnology-based drug delivery, this self-assembling Gem prodrug nanosystem constitutes a promising anticancer candidate.This study also offers new perspectives of the amphiphilic dendrimer nanoplatforms for the development of self-assembling prodrugs.
Self-assembly is a powerful approach for creating new functional materials in various applications [1].Of particular interest is the recently emerging self-assembling prodrug (SAPD) approach for drug delivery [2].SAPD is based on the spontaneous selfassembly of amphiphilic molecule into well-defined supramolecular structure, with the goal of enhancing drug efficacy while overcoming drug instability and toxicity.We report here our efforts to elaborate SAPDs for enhancing the stability and activity of the anticancer drug gemcitabine while reducing its toxicity.
Gemcitabine (Gem) is an important nucleoside drug widely used in clinical practice for treating various cancers including pancreatic, breast, ovarian and non-small cell lung cancers [3–8].However, the clinical performance of Gem is greatly compromised by its metabolic instability due to the fast enzymatic conversion to the inactive metabolite fluorodeoxyuridine (dFdU) [9].In addition,specific transporters, such as human equilibrative nucleoside transporter (hENT) [10], are required for the cellular intake of Gem; and the expression of such transporters is often low or suppressed in many cancer cells, resulting in an inefficient cellular uptake of Gem and hence poor treatment response.In order to achieve a satisfactory clinical response, high doses of Gem are usually administered and in combination with other drugs, which frequently lead to serious side effects.To overcome these limitations, various drug delivery systems have been developed for improving the therapeutic efficiency of Gem, with lipid and polymer vectors being the most widely studied [11–18].
However, as inert drug excipients and vector components account for the major part of the mass of such delivery systems, the drug loading is rather low.To solve this dilemma, the self-assembling prodrug (SAPD) approach represents a particularly promising strategy [2,19–21].SAPD incorporates drug that not only has a therapeutic purpose but also has a structural role within the resulting self-assembled nanoparticle.This effectively reduces the amount of drug carrier required and, at the same time, enables the exploitation of nanotechnology-based drug delivery, notably:(i) an abundance of drug molecules within the same nanoparticle, hence enhancing drug efficacy; (ii) the ability to enter cellsviaendocytosis, thus bypassing transporter-mediated cellular uptake; and (iii)the specific accumulation within tumorsviathe “enhanced permeability and retention” (EPR) effect for passive tumor targeting[22,23].
In this context, we have been particularly interested in elaborating nanotechnology-based self-assembling prodrugs of Gem.Inspired by the self-assembling dendrimer nanosystems established in our group for drug delivery (Fig.1A) [24–27], we developed amphiphilic dendrimers I and II as SAPDs of Gem (Fig.1B).Specifically, Gem entities are appended covalently to the terminals of the amphiphilic dendrimers, which are composed of a hydrophobic alkyl (C18) tail and a hydrophilic poly(amidoamine) (PAMAM) dendron.One of these Gem-dendrimer conjugates in particular, I, is able to self-assemble into small nanoparticles (GemNPs) that protect Gem against enzymatic transformation and promote effective cellular uptakeviaendocytosis while bypassing the transportermediated uptake and facilitating acid- and enzyme-promoted drug release, ultimately achieving much more potent anticancer activity with reduced adverse effects compared to the free drug Gem.We present these results below.
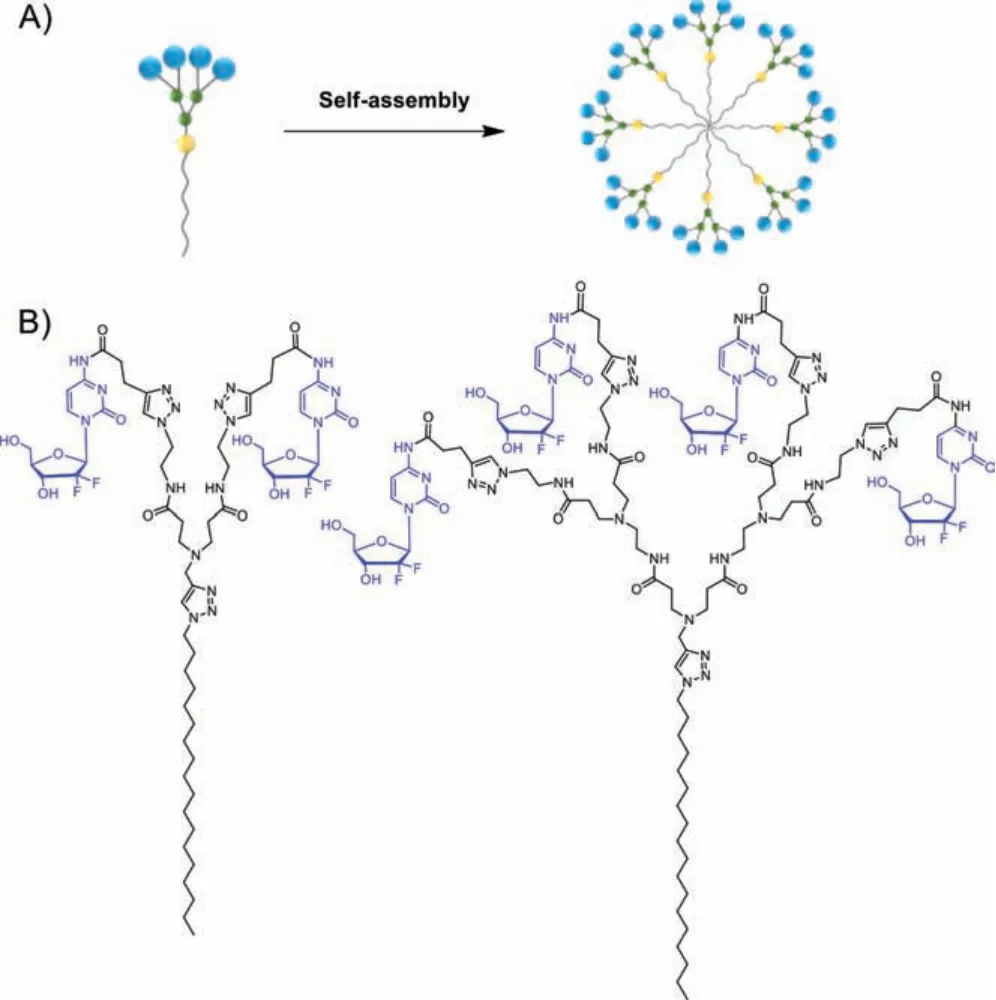
Fig.1.(A) Cartoon illustration of self-assembling of amphiphilic dendrimer into nanomicelle for drug delivery.(B) Chemical structures of the gemcitabine-dendrimer conjugates I and II in this work.
We synthesized the Gem-dendrimer conjugates I and IIviaclick chemistry using the alkynyl-bearing gemcitabine analogue III and the corresponding azido-carrying dendrimers 1 and 2, respectively (Scheme 1 and Schemes S1–S3 in Supporting information).The precursors III, 1 and 2 were prepared readily using reported protocols [28,29].The click reaction between III and 1 or 2 proceeded very well, and both I and II were obtained in pure state,as confirmed by NMR spectroscopy and high-resolution mass spectroscopy (HRMS) (Figs.S1–S4 in Supporting information).
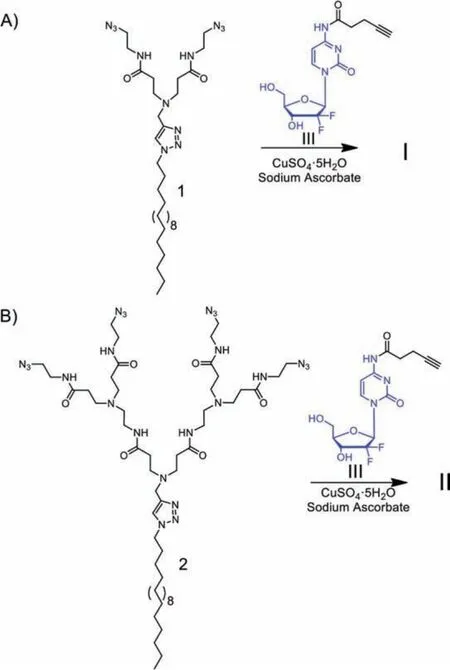
Scheme 1.Synthesis of the gemcitabine conjugated amphiphilic dendrimers I (A)and II (B) via click chemistry using the corresponding azido-carrying dendrimers 1 and 2, and the gemcitabine analogue III.
By virtue of the covalent prodrug conjugation, both I and II have high and stable loading content of Gem up to 40 wt% and 42 wt%, respectively, calculated from their molecular structure and chemical composition.Unfortunately, II was not soluble in water.We therefore continued all further studies using I, the conjugate showing much better solubility.
We first studied the self-assembly of I by determining its critical micelle concentration (CMC) using fluorescence spectroscopy with pyrene as fluorescent probe (Fig.S5 in Supporting information).The packing and self-assembling ability of I was demonstrated with a CMC value of 11 μmol/L.In addition, I formed spherical nanomicelles (hereafter referred to as GemNPs) of around 7.0 nm in water, as revealed by transmission electron microscopy(TEM) (Fig.2A).This was further confirmed with results obtained from dynamic light scattering (DLS) analysis, which clearly demonstrated the formation of small particles (Fig.2B) with dimensions in the range of typical nanomicelles.Importantly, the zeta potential of GemNPs, found to be around -20 mV (Fig.2C), enables the generation of electrostatic repulsion and prevents aggregation of NPs,making GemNPs colloidally stable.Also, the negatively charged NPs would be expected to demonstrate a repulsive interaction with the often negatively charged cell membrane and serum proteins, hence being less toxic.
In order to confirm the spontaneous self-assembling of I into GemNPs, we performed extensive runs (1.0 μs) of molecular dynamics (MD) simulations.Starting from a random distribution of I in solution, the MD approach permitted the achievement of stable GemNPs, as shown in Fig.2D.The calculated average micelle radius (Rmic) was around 3.4 nm (Fig.2E), matching the GemNPs dimensions obtained using both DLS and TEM.The electrostatic potential at the surface of the simulated GemNPs was found to be negative (Fig.2F), in agreement with the experimentally-derived negative surface zeta potential.Further examination of the conformational structures of the formed GemNPs and the relevant radial distribution of the amphiphilic dendrimer terminal groups (Fig.2G)revealed the absence of backfolding and the location of the Gem moieties at the micellar periphery, in line with the structure of the stable GemNPs shown in Fig.2D.
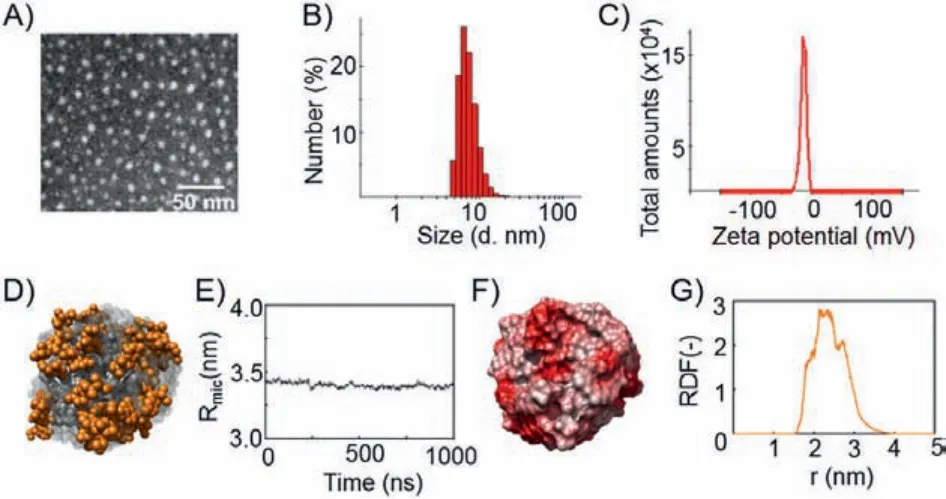
Fig.2.Self-assembling of the amphiphilic conjugate I into small and uniform nanomicelles (GemNPs).(A) Transmission electron microscopy (TEM), (B) dynamic light scattering (DLS) analysis, and (C) zeta potential measurement of the Gem-NPs.(D) Zoomed snapshot of a GemNP as extracted from the corresponding equilibrated molecular dynamics (MD) trajectory.All atoms are represented as spheres,with Gem moieties highlighted in orange (hydrogen atoms, water molecules, ions and counterions have been omitted for clarity).(E) Average micellar radius (Rmic)of the GemNPs along the equilibrated portion of the MD trajectory.(F) Electrostatic surface potential of the GemNP as extracted from MD simulations.(F) Electrostatic surface potential of the GemNP as extracted from MD simulations.(G) Radial distribution function of the Gem-bearing terminal groups as a function of the distance from the center of mass of the GemNPs.
Considering the metabolic instability of Gem due to its fast enzymatic deamination to the inactive form dFdU by cytidinedeaminase (CDA) that is responsible for the reduced efficacy of this anticancer drug [9], we wished to assess the protective capacity of GemNPs against this enzyme.We used anin vitroCDA ELISA assay to inspect whether CDA could metabolize the Gem entities within the GemNPs, with the free Gem serving as the control.Gem (100 μmol/L) or equivalents from GemNPs (100 μmol/L) were incubated with 0.25 μg of CDA at 37 °C, and the absorbance was recorded atλ= 280 nm.The decrease in the absorbance intensity was indicative of deamination [30].As shown in Fig.3A, free Gem was rapidly deaminated as illustrated by a significant reduction in its absorbance intensity in the presence of CDA.This result is consistent with the literature report showing the high susceptibility of free Gem to enzymatic deamination by CDA [30].However, conjugated Gem held within GemNPs was unaffected by CDA,as shown by the similar results obtained in the presence and in the absence of CDA.These results demonstrate that GemNPs effectively protected Gem from deamination.The resistance of GemNPs to CDA deamination can be explained by both the protection of the amino group in Gemviaamide bond formation in I and the steric hindrance of the Gem entities at the surface of the Gem-NPs (Fig.2D), the latter of which hampers access of CDA to Gem terminals.The GemNPs ultimately enhance therefore the metabolic stability of Gem.
Motivated by the increased metabolic stability and high drug loading offered by GemNPs, we further assessed the anticancer activity on different cancer cell lines including human pancreatic cancer cell lines PANC-1, Mia-PaCa-2 and SW1990 as well as the human breast cancer cell line MCF-7.Using CCK8 assay, we evaluated cancer cell proliferation upon treatment with GemNPs, the free drug Gem, the single Gem prodrug III and a non-treatment control (Fig.3B).The concentration of Gem was 50 μmol/L for all tested compounds.GemNPs demonstrated a much stronger ability to inhibit cancer cell proliferation with respect to the free Gem and the Gem prodrug III, highlighting the beneficial role of GemNPs in enhancing the antitumor activity of Gem.
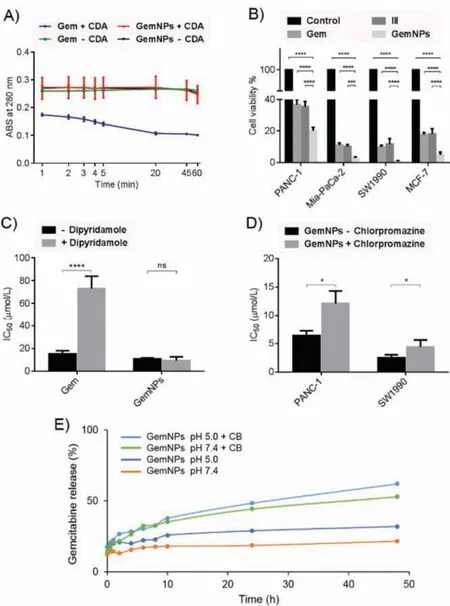
Fig.3.GemNPs are metabolically stable against cytidine deaminase (A), and effectively inhibit cancer cell proliferation (B) in a nucleoside transporter-independent manner (C), benefiting endocytosis-based cellular uptake (D), and acid- and enzyme-promoted drug release (E).*P <0.05; ***P <0.001; ****P <0.0001.
A reasonable explanation for the observed enhanced antiproliferative activity of GemNPs compared to the free drug Gem is an increased amount of Gem molecules able to enter cancer cells through alternative cell entry pathways.Indeed, cellular uptake of hydrophilic Gem requires special transport systems such as the human equilibrative nucleoside transporter (hENT) [10], whereas nanosized particles enter cells oftenviaendocytosis.In order to check whether cellular uptake of GemNPs occurs independently of hENT, we assessed and compared the antiproliferative activity of GemNPs in the presence and absence of dipyridamole, a hENT inhibitor.Our results show that GemNPs demonstrated similar antiproliferative activity in the presence and absence of dipyridamole, with an IC50value of 11 μmol/L and 9.4 μmol/L, respectively (Fig.3C).This indicates that blocking the nucleoside transporter did not affect the cytotoxicity of GemNPs.As a control,the IC50values for free Gem were 73 μmol/L and 15 μmol/L in the presence and absence of dipyridamole, respectively (Fig.3C),highlighting that the cytotoxicity of free Gem is highly reliant on the nucleoside transporter, in accordance with the literature report[31].Collectively, our results demonstrate that GemNPs inhibit cancer cell proliferation in a nucleoside transporter-independent manner.
In order to confirm that endocytosis is indeed the cellular uptake pathway used by GemNPs, we further evaluated and compared the antiproliferative activity of GemNPs in the presence and absence of chlorpromazine, an inhibitor for endocytosis pathway on the pancreatic cancer cells PANC-1 and SW1990.As shown in Fig.3D, GemNPs exhibited much stronger antiproliferative activity in the absence of chlorpromazine on both PANC-1 and SW1990 cell lines.These data demonstrate that blocking endocytosis effectively reduced the anticancer activity of GemNPs, indicating that endocytosis is importantly involved in cellular uptake of GemNPs.
It is to mention that the endocytosis-mediated cell uptake can further favor the acid- and enzyme-promoted drug release of the prodrug nanoparticles in the acidic and protease-rich endosomes.This is particularly beneficial to drug release from GemNPs, as Gem is covalently linked within Iviaamide linkage and reluctant to be released under normal physiological conditions.Usually,the amide bond is very stable, and needs specific enzymatic action to be cleaved under mild conditions, hence being frequently used as an enzyme-responsive linker of gemcitabine and other drug molecules [2,32].Nevertheless, the exocyclic amide bond which protects the amino function in gemcitabine is often activated for breakdown thanks to the electron withdrawing character of the nucleobase.We therefore assessed Gem release from GemNPs at pH 5.0 and in the presence of the enzyme, cathepsin B (CB), in order to mimic the acidic and enzyme-rich endosomes.Cathepsin B is a cysteine protease which plays an prominent role in intracellular proteolysis, and has been frequently used as a model enzyme for studying enzyme-promoted drug release from prodrug systems[33].Solutions of GemNPs (0.50 mg/mL, 0.38 mmol/L, equivalent to 0.76 mmol/L Gem) were incubated in buffer at pH 7.4 or pH 5.0 and in the presence and absence of 20 μL of Cathepsin B(2445 U/mg) at 37 °C, and the released Gem was quantified using HPLC.As shown in Fig.3E, Gem release was increased at pH 5.0,and significantly enhanced in the presence of CB when compared to that at neutral pH 7.4 and in the absence of enzyme.Specifically, the drug release attained more than 60% of the total Gem in the presence of CB and at pH 5.0 after 48 h.This result highlights a pH- and enzyme-promoted release of Gem from the GemNPs, being both effective and sustainable.
Encouraged by all these promising features of GemNPs, we wanted to evaluate their anticancer activityin vivousing pancreatic tumor-xenograft mice.Pancreatic cancer is one of the most lethal and devastating human cancers; and it has an overall survival rate less than 10% and a median survival less than 6 months[5].Although Gem is the standard-of-care chemotherapy for treating pancreatic cancer, the therapeutic outcomes remain largely unsatisfactory because of the specific metabolism and transportation mechanisms of Gem [34].Consequently, there is a pressing need to explore novel therapeutic modalities to combat pancreatic cancer.In this study, we therefore wanted to assess GemNPs for the anticancer efficacy using pancreatic cancer as the cancer model.
Before starting the animal experiments, we first assessed the potential hemolytic toxicity of GemNPs using mouse red blood cells.Gratifyingly, no notable hemolysis was observed with red blood cells after treated with GemNPs at varying concentrations from 0.50 μmol/L to 42 μmol/L, using water as a positive control and PBS buffer as a negative control (Fig.4A).We then assessed the anticancer performance of GemNPsin vivousing pancreatic cancer SW1990 xenograft mice treated with saline, Gem or GemNPs at the equivalent Gem dose.We set the dosage of Gem of 7.0 mg/kg on the basis of the abovein vitroevaluation data and numerous reported studies [35–37] in the view to inspecting the efficacy and the safety profile of GemNPs.The tumor size and the mouse body weight were monitored over 3 weeks.As shown in Fig.4B, GemNPs significantly attenuated tumor growth, compared with the control groups of mice treated with saline or Gem alone,underlining the superior anticancer activity of GemNPs compared to the parent drug Gem.In addition, no significant body weight loss was observed upon treatment with GemNPs (Fig.3C), suggesting that GemNPs were well tolerated.
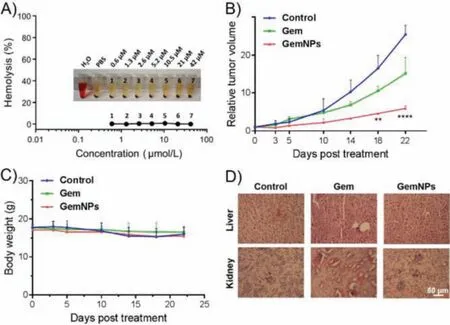
Fig.4.GemNPs show safe and effective antitumor activity.(A) Hemolysis assay of GemNPs.(B) Effective antitumor activity of GemNPs in pancreatic cancer SW1990 xenograft nude mice.(C) The body weight of the mice was recorded throughout the whole treatment period.(D) In vivo toxicity assessment of GemNPs and Gem alone was performed by sectioning and H&E staining of liver and kidney. n = 3 or 4, **P <0.01; ****P<0.0001.
As Gem is reported to cause serious side effects such as nephrotoxicity and hepatotoxicity, we further assessed the toxicity of GemNPs using H&E staining to assess any pathological changes in both liver and kidney of mice treated with GemNPs and free Gem intravenously.Mice treated with GemNPs showed no discernible effects of toxicity in either of these two organs (Fig.4D).In contrast, however, we observed acute tissue injuries in both liver and kidney of xenograft mice treated with free Gem.These results emphasize the safety of GemNPs compared with free Gem, and collectively demonstrate the potential of this Gem prodrug for future therapeutic implementation.
In summary, we developed amphiphilic dendrimer conjugates I and II as self-assembling gemcitabine prodrugs.The conjugate I is particularly promising because of its high drug loading (40%) and self-assembling ability into small and stable nanoparticles (Gem-NPs).GemNPs significantly increased the metabolic stability of Gem by preventing enzyme degradation, and at the same time, effectively enhanced cellular uptakeviaendocytosis while bypassing nucleoside transporter-mediated uptake.In addition, GemNPs released Gem effectively and sustainably in response to acid and enzyme action, leading to effective antiproliferative activity on different cancer cells and in tumor-xenograft mouse model without adverse effects.Collectively, these results demonstrate that this selfassembling Gem prodrug nanosystem constitutes a promising anticancer candidate.This study has also underlined the potential of self-assembling prodrug nanosystem to harness the combined advantages of both prodrug and nanotechnology-based drug delivery for improving drug stability and efficacy as well as reducing drug toxicity.We are actively pursuing this research direction.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Financial support was from the National Natural Science Foundation of China (Nos.81903567, 31600109), Henan Programs for Science and Technology Development (No.182102310221), Xinxiang Innovative Technology Team (No.CXTD17004), the PhD startup fund of Xinxiang Medical University (No.505158) and the Ligue Nationale Contre le Cancer.This publication is based upon work from COST Action CA 17140 "Cancer Nanomedicine from the Bench to the Bedside".
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.083.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction