
Construction of a low-valent thiolate-bridged dicobalt platform and its reactivity toward hydrogen activation and evolution
2022-06-20 07:59TaoMeiDaweiYangLinanSuBaominWangJingpingQu
Chinese Chemical Letters 2022年5期
Tao Mei, Dawei Yang, Linan Su, Baomin Wang, Jingping Qu
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, China
Keywords:Low-valence Thiolate-bridged Cobalt Homolytic H2 cleavage Hydride Hydrogen evolution
ABSTRACT Low-valence transition metallic complexes have drawn longstanding attention due to their high reactivity toward catalytic transformation of various small molecules.Among these known complexes, the lowvalence metal centres are commonly stabilized by neutral bulky ligands with strong electron-donating capacity.However, low-valence bimetallic complexes supported by anionic sulfur and cyclopentadienyl ligands are still difficult to obtain in high isolated yield.Herein, we report the synthesis and characterization of two scarce thiolate-bridged CoICoII and CoICoI complexes bearing sterically demanding ligands through two stepwise one-electron reduction processes.Interestingly, the CoICoII complex can facilely promote the homolytic cleavage of dihydrogen across the short Co-Co metallic bond to give a CoIICoIII dihydride bridged complex, which is capable of serving as a competent hydrogen atom transfer agent.Moreover, the anionic CoICoI complex can trigger a stepwise hydrogen generation cycle involving several isolated and structurally well-characterized intermediates.
Low-valent transition metal complexes commonly exhibit high reactivity toward small molecule activation and further functionalization [1–3].However, this kind of complex always has a tendency to undergo disproportionation into more stable metallic complexes with higher oxidation state and zero-valent metal atoms [4].To stablize the highly active metal center, strong electron-donating neutral bulky ligands such as phosphines and carbenes were usually adopted [1,5].In comparison, anionic ligands are not benefical for the construction of low-valent metallic complexes, for example, cyclopentadienyl or weak-field sulfur donors.Especially, lowvalent thiolate-bridged binuclear complexes are rarely reported to date and their potential reactivities are also poorly explored.
In nature, widely distributed hydrogenases commonly adopt thiolate-bridged bimetallic core frameworks for hydrogen activation and evolution [6,7].Notably, the low-valence species (FeIFeIor FeIINiI) are generally regarded as the trigger for the corresponding catalytic transformation cycles.Inspired by biological systems,construction of suitable low-valence binuclear platforms based on earth-abundant metals for hydrogen activation and production is desirable and challenging.
As an inexpensive and earth-abundant metal with rich redox behavior, cobalt has been increasingly attractive in the development of efficient catalysts for hydrogen activation and production.In recent two decades, a variety of synthetic cobalt complexes were proven to be good promoters for catalytic hydrogen oxidation and generation [8–10].In comparison with frequently reported mononuclear cobalt complexes, binuclear cobalt complexes are very limited [11–18].For example, Fukuzumiet al.synthesized a dicobalt hydride complex supported by multidentate nitrogen ligands that can undergo the heterolytic cleavage of the CoIII-H bond by proton to release hydrogen [12].Recently, a scarce thiolate-bridged dicobalt azido complex [Cp*CoIII(μ-SEt)2(μ1,1-η1:η1-N3)CoIIICp*][BPh4] (Cp*=η5-C5Me5) was reported by our group, which can achieve an uncommon homolytic cleavage of dihydrogen across the bridging azido group [17].However, among these dicobalt complexes, there remains no example that can not only achieve hydrogen evolution, but also hydrogen activation.To our knowledge, there are also only a handful of other metal complexes with similar reactivity, but the embodiment of bifunctional feature usually needs electrochemical assistance [19–21].
In our previous work, we designed and synthesized a series of new thiolate-bridged bimetallic complexes, which can mediate the chemical simulation of nitrogenase [22–24] or small molecule activation and catalytic conversion [25–28].As an important supplement and extension of biomimetic diiron cooperative systems,we also constructed thiolate-bridged dicobalt functional platform,which exhibits versatile reactivities [15,17,18,29,30].In this paper,the bulky monodentate adamantyl (Ad) thiolate was adopted as a bridging ligand for constructing new dicobalt functional systems.This choice is mainly based on the following two considerations:(1) its steric hindrance effect that is beneficial to the formation of the low-coordinate metal centres; (2) its electron-donating capacity that can stabilize the low-valent metal centres.Following this line, we successfully construct a series of novel low-valent thiolatebridged dicobalt complexes.More importantly, this {Co(μ-SAd)Co}scaffold developed here can not only facilely mediate a homolytic cleavage of H2across the short metal-metal bond to generate a scarce dihydride species, but can also promote H2evolution following a stepwise process evidenced by the isolation of all potential key intermediates.
Our study commenced with treatment of precursor [Cp′Co(μ-Br)2CoCp′] (Cp′ =η5-1,2,4-tBu3C5H2) [31] with 1 equiv.of NaSAd from –78 °C to room temperature, affording a thiolate-bridged formal CoIICoIIcomplex [Cp′Co(μ-SAd)(μ-Br)CoCp′] (1) in 74% yield(Scheme 1).In the proton nuclear magnetic resonance (1H NMR)spectrum of 1, two paramagnetically broadened peaks appear atδ5.10 and 6.23 ppm, which were assigned totBu groups in the Cp′ ligands.The other signals falling in the range of 1.05–1.61 ppm belong to the adamantyl group.Moreover, effective magnetic moment (μeff) of 1 at room temperature was determined to be 2.53μBusing Evans’method [32], which suggests the overall spin stateS= 1.
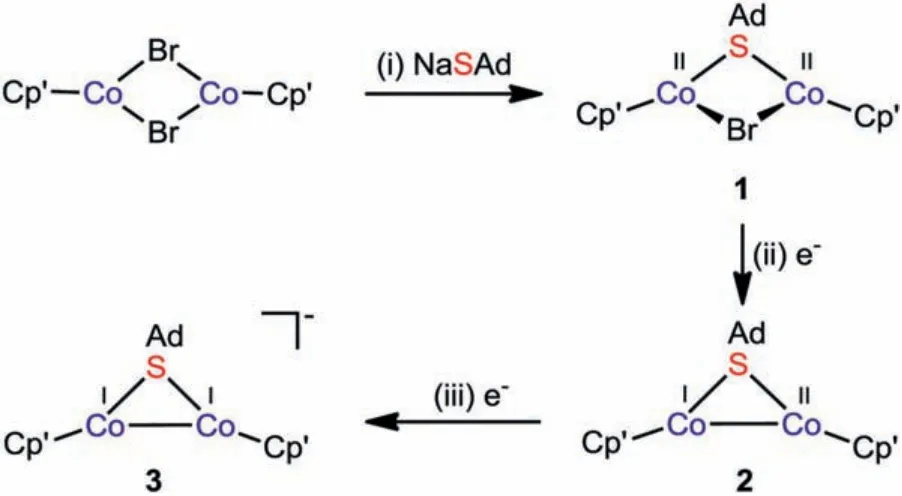
Scheme 1.The synthetic route of low-valence thiolate-bridged dicobalt complexes.Reagents and conditions:(i) 1 equiv.NaSAd, THF, -78 °C to r.t., 74%; (ii) 1 equiv.KC8, THF, -78 °C to r.t., 88%; (iii) 1 equiv.KC8, 1 equiv.crypt-222, THF, -78 °C to r.t., 58%.
The molecular structure of 1 was confirmed by X-ray diffraction analysis (Fig.1a).The overall framework consists of two {Cp′Co}subunits, which are bridged by one adamantyl thiolate and one bromide ligand.The distance between the two cobalt centres of 2.9491(9) Å in 1 is remarkably longer than those of 2.36–2.68 Å for other thiolate-bridged CoIICoIIcomplexes [Cp*Co(μ-SR)2CoCp*](R =iPr, Ph) [18].This is presumably attributed to the steric congestion of bulkytBu and bromine.In addition, the dihedral angle of the two Cp′ rings is 43.029(0)°, which can provide enough space for small molecule activation.
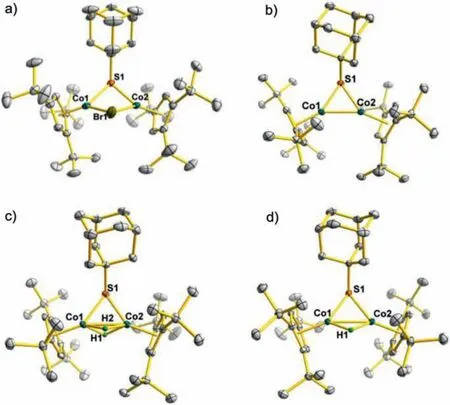
Fig.1.ORTEP (ellipsoids at a 50% probability) diagrams of complexes 1 (a), 2 (b),4 (c) and 5 (d).The hydrogen atoms except for bridging hydrides are omitted for clarity.
Considering the rich valence change of cobalt, the redox property of complex 1 was next investigated by cyclic voltammetry.As shown in Fig.2, the cyclic voltammogram of 1 exhibits a quasireversible reduction event atE1/2red= -1.66 V and an irreversible one atEpred= -2.01 V (all potentials areversusferrocene+/0),which are assigned to CoIICoII/CoIICoIand CoIICoI/CoICoIredox couples, respectively.The above electrochemical measurement implies CoIICoIIcomplex 1 may be further reduced to generate lower valence species.Hence, we subsequently examined the possibility of the chemical reduction of 1 (Scheme 1).Treatment of CoIICoIIcomplex 1 with 1 equiv.of potassium graphite (KC8) resulted in a one-electron reduction to produce [Cp′CoI(μ-SAd)CoIICp′] (2)in 68% yield.Notably, in the presence of 2.2.2-cryptand (crypt-222), CoIICoIcomplex 2 can be further reduced by KC8to afford[Cp′CoI(μ-SAd)CoICp′][K(crypt-222)] (3), which represents the first example of thiolate-bridged formal CoICoIcomplex.Surprisingly,this scarce electron-rich CoICoIcomplex is stable at room temperature and does not undergo the common disproportionation into more stable species, which may be attributed to the protection effect of sterically bulky ligands [4].
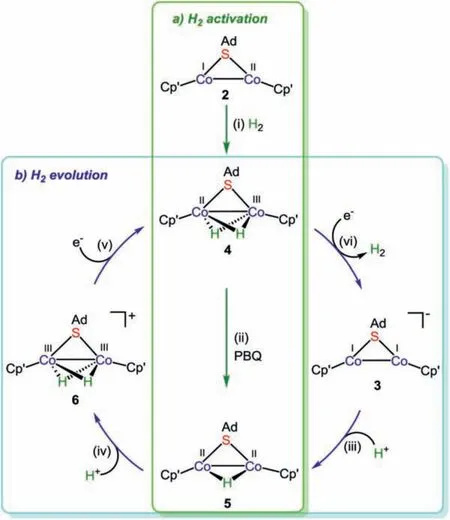
Scheme 2.The transformation pathways of hydrogen activation and evolution promoted by low-valence dicobalt complexes.Reagents and conditions:(i) H2 (1 atm),THF, -78 °C to r.t., 86%; (ii) 1 equiv. p-benzoquinone (PBQ), THF, -78 °C to r.t., 69%for 5, and 65% for hydroquinone; (iii) 1 equiv.LutH·BArF4, Et2O, r.t., 2 h, 39%; (iv)1 equiv.LutH·BArF4, THF, r.t., 4 h, 79%; (v) 1 equiv.CoCp2, THF, r.t., 4 h, 68%; (vi) 1 equiv.KC8, 1 equiv.crypt-222, THF, -78 °C to r.t., 41% for 3, and 81% for H2.
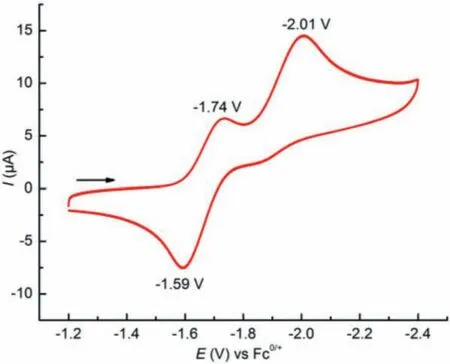
Fig.2.Cyclic voltammogram of 1 in THF at 100 mV/s with 0.1 mol/L nBu4NPF6 as supporting electrolyte and the potentials are referenced versus Fc+/0.
Complex 2 should also be a paramagnetic species supported by1H NMR spectroscopy.As expected, there are three broadened peaks with paramagnetic shift for Cp′ ligands atδ-6.10, -2.38 and -1.93 ppm in the high field.Furthermore, the solution magnetic measurement of 1.93μBfor 2 is in good agreement with the overallS= 1/2 supported by density functional theory (DFT)calculations (Table S10 in Supporting information).Differently, the1H NMR spectrum of 3 exhibits several intense resonances in the common region of 1–2 ppm for methyl and methylene protons,which are attributed to the Cp′ and SAd groups.The molecular structure of 2 closely resembles the anionic part of 3 (Fig.1b).Their core frameworks both consist of an unprecedented {Co(μ-SAd)Co} structural subunit.The Co–Co bond length of 2.3663(3) Å in 2 is very short, which suggests the existence of strong metalmetal bonding interaction between the two cobalt centres.Upon reduction, the Co–Co bond distance in 3 is remarkably elongated to 2.5241(5) Å.
With two low-valent coordinatively unsaturated dicobalt complexes in hand, we subsequently examined their reactivity toward small molecule activation and transformation.Unexpectedly, under 1 atmosphere of H2, complex 2 can facilely promote the homolytic cleavage of H2at room temperature to give a scarce dicobalt dihydride-bridged complex [Cp′Co(μ-SAd)(μ-H)2CoCp′] (4)in high yield (Scheme 2).Owing to the occurrence of the oxidative addition of H2, the formal valences of the two cobalt centres varied from CoICoIIin 2 to CoIICoIIIin 4.There is no diagnostic signal for the bridging hydrides observed in the high field region of the1H NMR spectrum of 4, which should be attributed to the paramagnetic influence [33,34].
To provide more evidence for the existence of two bridging hydrides, we conducted the electrospray ionization-high resolution mass spectrometry (ESI-HRMS) analysis.Gratifyingly, an anticipated molecular ion peak withm/z753.4250 (calcd.753.4254) was detected.Furthermore, we performed the isotope labeling experiment by replacing H2with D2to give the corresponding [Cp′Co(μ-SAd)(μ-D)2CoCp′] (D-4).The ESI-HRMS analysis of D-4 clearly exhibits a molecular ion peak withm/z755.4405 (calcd.755.4379),which has a mass shift of +2 compared to its unlabeled congener(Fig.S21 in Supporting information).These experiments not only confirmed the presence of two hydrides but also verify that they are originated from H2.
Because hydrides can serve as reductive equivalents for unsaturated substrates, we probed whether dihydride species 4 is a good H-atom donor or not.Upon treatment of 4 with stoichiometricp-benzoquinone (BDFEO–H= 72.6 kcal/mol in DMSO) [35],one bridging hydride underwent a hydrogen atom transfer process to afford hydroquinone, accompanied with the formation of dicobalt monohydride-bridged complex [Cp′Co(μ-SAd)(μ-H)CoCp′](5).Similar to 4, there is also no diagnostic signal for the bridging hydride in the1H NMR spectrum of 5.Unexpectedly, the bridging hydride in 5 is inert and cannot interact with various unsaturated substrates.
Crystallographic analysis unambiguously confirmed the molecular structures of 4 and 5 as shown in Figs.1c and d.Two complexes both contain a robust {Co(μ-SAd)Co} framework as precursor 2.The Co–Co distance of 2.4135(5) Å in 4 is slightly shorter than that of 2.4514(4) Å in 5.The bridging hydrides in 4 and 5 were located in the difference map and their positions and isotropic displacement parameters were refined without restraints.Furthermore, DFT calculations provide good agreement with the Co–Co distances as well as the Co–H distances in 4 and 5, allowing for the inherent uncertainty in the H-atom positions (Figs.S35 and S36 in Supporting information) [36].
Although proton reduction to H2promoted by thiolate-bridged bimetallic complexes has been extensively investigated [37–40],a complete transformation cycle containing a series of structurally characterized intermediates involved in every elemental step remains rarely reported.In this regard, we attempted to probe a potential transformation route to hydrogen evolution on this dicobalt scaffold.The CoICoIcomplex 3 readily undergoes protonation by LutH·BArF4(Lut = 2,6-lutidine, BArF4= B(3,5-(CF3)2C6H3)4), affording an oxidative addition product, CoIICoIImonohydride-bridged complex 5 in moderate yield.Unexpectedly,complex 5 can further react with 1 equiv.of LutH·BArF4to generate a rare CoIIICoIIIdihydride-bridged complex [Cp′Co(μ-SAd)(μ-H)2CoCp′][BArF4] (6) in 79% yield.The1H NMR spectrum of 6 provides further evidence for the existence of two bridging hydrides, wherein two diagnostic resonances appear atδ-20.70 and-20.17 ppm.Compared with known dicobalt hydride bridged complexes [15,18,41], these two resonances for the bridging hydrides appear in the higher field region.Further structural characterization reveals the cationic part of 6 resembles the neutral dicobalt dihydride-bridged 4 (Fig.S6 in Supporting information).Complex 3 represents a rare bimetallic complex that can undergo double protonation to generate dihydride species instead of hydrogen evolution [42,43].
To get one step further, chemical reduction of 6 was carried out using CoCp2as a reductant.The product was verified as neutral dihydride-bridged complex 4 by spectroscopic analyses.Reversibly,neutral dihydride-bridged complex 4 can be also one-electron oxidized by Fc·BArF4to regenerate 6.Notably, further reduction of complex 4 can be achieved by using stronger reductant KC8in the presence of crypt-222 to readily afford 3 in moderate yield.Meanwhile, the headspace analysis by gas chromatography (GC) corroborated H2(Fig.S30 in Supporting information) was produced from the two bridging hydrides in 81% yield during the reduction process of 4.
Finally, we also examined the catalytic reactivity of hydrogen evolution by cyclic voltammetry and the key intermediate dicobalt dihydride 4 was chosen as a representative target.Using trifluoroacetic acid (TFA,pKaMeCN= 12.65) [44] as proton sources, complex 4 can serve as an electrocatalyst with reduction peak potential at -2.5 Vvs.Fc+/0for proton reduction to hydrogen evolution (Fig.S24 in Supporting information).The ratio of the catalytic current(ic) to reduction peak current (ip) in the presence of 10 mmol/L TFA was calculated to be 6.36, which is comparable to other thiolatebridged dicobalt complexes [15,18], but less efficient than some other thiolate-bridged bimetallic complexes [45–49].Additionally,the further performance of the bulk electrolysis in the presence of excess TFA at –2.8 V (Fig.S28 in Supporting information) and a series of control experiments such as rinse test [50,51] (Fig.S29 in Supporting information) provided more robust evidence that the molecular dicobalt complex promotes the catalytic hydrogen evolution.
In summary, we adopt the sterically demanding ligands to synthesize a series of rare low-valence thiolate-bridged dicobalt complexes.The CoICoIIcomplex can facilely realize the homolytic cleavage of dihydrogen across the short Co-Co bond to give a scarce CoIICoIIIdihydride bridged complex, which can serve as a competent hydrogen atom transfer agent.Interestingly, its anionic CoICoIanalogue exhibits a strong affinity to proton and triggers the hydrogen generation through two successive protonation processes following two one-electron reduction events.Notably, all key intermediates during this transformation process are isolated and welldefined.This [Co(μ-SAd)Co] system gives a new opportunity for the development of low-valence bimetallic catalysts.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos.21690064, 22001031), Key Laboratory of Biobased Chemicals of Liaoning Province of China, the “111” project of the Ministry of Education of China and the Fundamental Research Funds for the Central Universities (No.DUT19RC(3)013).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.014.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction