
Pincer iridium(III)-catalyzed enantioselective C(sp3)-H functionalization via carbenoid C–H insertion of 3-diazooxindoles with 1,4-cyclohexadiene
2022-06-20 07:59NanLiXiaoyanYangYanyanZhuFangWangJunfangGongMaopingSong
Chinese Chemical Letters 2022年5期
Nan Li, Xiaoyan Yang, Yanyan Zhu, Fang Wang, Junfang Gong, Maoping Song
College of Chemistry, Green Catalysis Center, Zhengzhou University, Zhengzhou 450001, China
Keywords:Asymmetric catalysis C–H functionalization Pincer iridium(III) catalyst Carbenoid C–H insertion 3-Diazooxindole Chiral 3-substituted oxindole
ABSTRACT The asymmetric carbenoid C–H insertion of 3-diazooxindoles into 1,4-cyclohexadiene has been accomplished in the presence of chiral bis(imidazoline) NCN pincer iridium(III) complexes as the catalysts.With a catalyst loading of 0.5 mol%, the reactions proceeded smoothly at 0 °C to afford a variety of chiral 3-substituted oxindoles in good yields with moderate to excellent enantioselectivities (up to 99% ee).The protocol exhibits good functional group tolerance with respect to 3-diazooxindoles and is readily scaled up to 2 mmol scale without any loss in activity and enantioselectivity.Density functional theory (DFT)calculations have been performed to better understand the reaction mechanism and to explain the stereochemical outcome of the reactions.
Catalytic enantioselective insertion reaction of metal carbenoids, generatedin situfrom metal mediated decomposition of diazo compounds, into C–H bonds represents an important and powerful strategy for C–H bond functionalization and construction of C–C bonds [1–4].In particular, chiral dirhodium(II) complexes catalyzed intermolecular C–H insertions of donor/acceptorsubstituted carbenoids have been extensively investigated and found broad applications in the synthesis of natural products, pharmaceuticals, bioactive compounds and other complex chiral targets[5,6].Successful dirhodium(II) catalysts include, among others, binaphtholphosphates,ortho-metalated arylphosphine dirhodium, especially various dirhodium carboxylates and carboxamidates complexes.Undoubtedly, rhodium complexes have been widely established as the most effective and versatile catalysts for asymmetric carbenoid C–H bond insertion reactions.In contrast, only a limited number of iridium complexes have been reported as viable catalysts for such reactions despite the fact that less expensive Ir belongs to the same group as Rh [7].Compared with their rhodium counterparts, iridium carbenoids tend to have a lower electrophilic character due to the increased metal to ligand backbonding, which makes the iridium carbenoids less reactive but more selective [6,7].Indeed, researches from Suematsu and Katsuki[8], Weldyet al.[9] have demonstrated the unique reactivity of iridium carbenoids by means of which some challenges in the area have been addressed.In their works, intermolecular C–H insertions ofα-diazopropionates [8] and ethyl diazoacetates [9] were successfully realized with high enantioselectivities (83% ~>99%eeand up to 96%ee, respectively) for the first time by using chiral Ir(III)-(salen) or bis(imidazoline) pincer Ir(III) complexes as catalysts.It is well known that the reactions with these two types of diazo compounds are quite challenging.Forα-diazopropionates, they are inclined to undergo competitiveβ-hydride elimination.While for ethyl diazoacetates, controlling site, chemo- and stereo-selectivity of the reactions is difficult owing to the very high reactivity and non-prochiral property of the corresponding acceptor-only metallocarbenoids.
Oxindole derivatives, including chiral 3-substituted oxindoles,are widely found in alkaloid natural products, pharmaceuticals and synthetic biologically active molecules [10,11].They can also serve as versatile precursors in organic synthesis and as central intermediates in the synthesis of a number of naturally occurring compounds [12,13].Transition metal catalyzed intermolecular carbenoid C–H insertion of 3-diazooxindoles, a kind of cyclic diazoamide and also a donor-acceptor diazo compound,provides a direct and efficient approach for the construction of 3-substituted oxindoles.Thus, various achiral 3-substituted oxindoles with diverse structures have been successfully prepared through Rh, In, Au or Ru catalyzed non-asymmetric C–H insertion of 3-diazooxindoles with a wide range of substrates such as indoles, pyrroles, anthracenes, carbazoles,β-enaminoesters,N,Ndisubstituted anilines, phenols,β-enaminones [14–24].However, to the best of our knowledge, there are no reports on enantioselective C–H carbenoid insertion of 3-diazooxindoles for the synthesis of chiral 3-substituted oxindoles.
We have been interested in exploring applications of pincer metal complexes in metal-catalyzed transformations including those of chiral ones in enantioselective reactions [25–28].Very recently, we found that pincer Ir(III) complex 1d with a chiral bis(imidazolinyl)phenyl ligand (abbreviated as Phebim, Fig.1) exhibited good activity and stereocontrol (up to 86%ee) in asymmetric carbenoid C–H insertion ofα-aryl-α-diazoacetates withNprotected indoles [29].To develop more Ir-catalyzed, especially pincer Ir-catalyzed intermolecular carbenoid C–H insertions for further exploring and understanding the chiral iridium chemistry in the area, we herein present the use of the pincer (Phebim)Ir complexes (Fig.1) in catalytic enantioselective C–H insertion of 3-diazooxindoles into 1,4-cyclohexadiene.The current work represents the first example of asymmetric intermolecular C–H carbenoid insertion reaction using 3-diazooxindoles as carbenoid precursors.
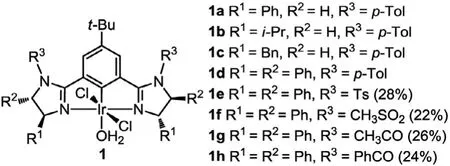
Fig.1.Chiral bis(imidazoline) NCN pincer iridium(III) complexes.
In our previous study, the pincer (Phebim)Ir complex 1d with a catalyst loading of 3 mol% gave the best result among complexes 1a-1d [29].For further catalyst optimization, synthesis of the new (Phebim)Ir complexes 1e-h were carried out (for details see Supporting information).In comparison with 1d, complexes 1e-1h have the same (4S,5S)-diphenyl substituents but differentNsubstituent on the imidazoline ring.Synthetically, a chiral diamine was used as the chiral source for complexes 1e-1h, while for complex 1d (and also complexes 1a-1c) the chiral substituents originated from chiral amino alcohols.In addition, it was found that the introduction ofN-electron withdrawing group (N-sulfonyl or acyl) on the imidazoline ring was detrimental to the direct metallation of the corresponding Phebim-H ligands, leading to the obviously lower yields of complexes 1e-1h (22%-28%) in this step when compared with 1d (46%) and 1a-1c (34%-45%).X-ray single crystal analysis of the complex 1h confirmed that it is the expected H2O-bound six-coordinate pincer complex with meridionally tridentate coordination of the Phebim ligand (Fig.2, CCDC:1971477).
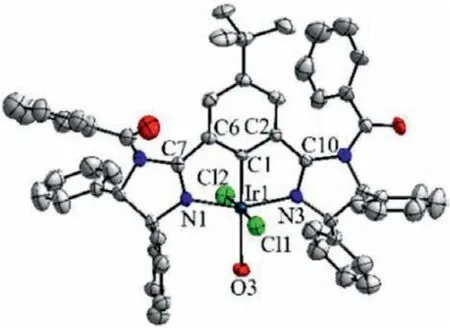
Fig.2.Molecular structure of complex 1h.
The C–H insertion of 3-diazo-1-methylindolin-2-one with 1,4-cyclohexadiene was first chosen as a model to evaluate the potential of the obtained (Phebim)Ir complexes (for details see Supporting information).Encouragingly, except for complexes 1b and 1c,all other complexes including 1a and 1d-1h could give the desired product 3a in high yields (81%-97%) with excellent enantioselectivities (93%-97%ee) when the reactions were conducted with a large excess of the cyclodiene (21.5 equiv.) under solvent-free condition at room temperature with a catalyst loading of 1.0 mol% for 12 h (Table 1, entries 1–6).Considering that the synthetic yield of 1a was higher than those of 1f-1h in the metallation step (44%vs.22%-26%), complex 1a was then further utilized as the catalyst for optimization of reaction conditions.A screening of temperature indicated that in the range of -20~40 °C lowering the temperature helped to improve the yield of product 3a by inhibiting side reactions such as dimerization of the diazo compound and 0 °C was found to be the optimal temperature (entry 7).Next, we tried to reduce the amount of 1,4-cyclohexadiene and the catalyst loading.It turned out that the use of 10.8 equiv.of the cyclodiene in the presence of CH2Cl2solvent (0.2 mL) and with a catalyst loading of 0.5 mol% could also afford a good result (entry 8, 88% yield with 93%ee).Pleasingly, under these conditions, complex 1g gave a better result than 1a in terms of both yield and enantioselectivity (entry 9, 93% yield with 97%ee).Increasing the CH2Cl2solvent volume resulted in decreased yields and/or enantioselectivity of 3a.Based on the results and also for convenience of manipulation, 0.5 mL of CH2Cl2as the solvent was considered to be appropriate (entry 10).Finally, TLC monitoring of the reaction showed that the diazo compound was completely consumed within 8 h, furnishing the product 3a in 87% yield with 96%ee(entry 11).
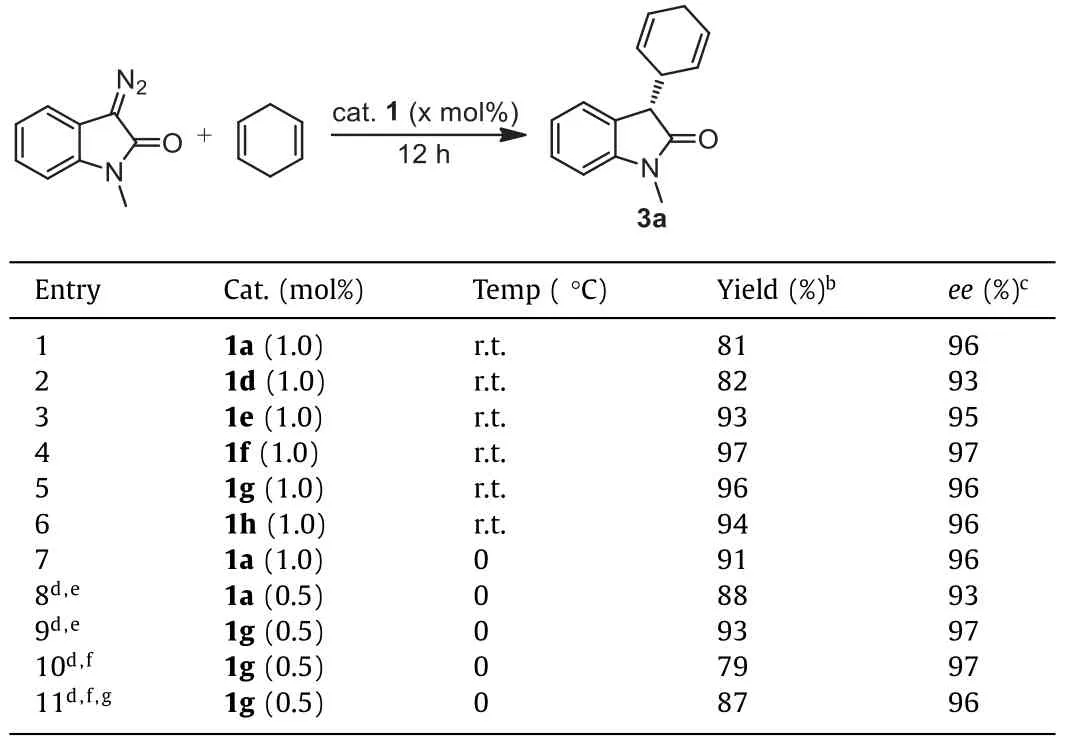
Table 1 Optimization of reaction conditions.a
The substrate scope of 3-diazooxindoles was then investigated under the optimized conditions.As shown in Table 2,various substituted 3-diazooxindoles reacted smoothly with 1,4-cyclohexadiene to furnish structurally diverse oxindoles 3 bearing a chiral cyclohexa-2,5–dien-1-yl group at the 3-position in moderate to excellent yields (42%-99%) with moderate to excellent enantioselectivities (51%-99%ee).The substituents involve electronwithdrawing groups (EWG) including F, Cl, Br, I, CF3, and OCF3as well as electron-donating groups (EDG) including Me and OMewhich are located at 4-, 5-, 6- or 7-position of 3-diazooxindoles.In general, 3-diazooxindoles containing an EDG at 5-, 6- or 7-position reacted quite well with the cyclodiene, giving the corresponding products 3 in high yields (76%-95%) with invariably excellent enantioselectivities (entries 4–6, 13, 14, 18 and 19, 94%-99%ee).In contrast, 3-diazooxindoles with an EWG exhibited lower reactivity and an extended reaction time (24vs.8 h) was needed to ensure satisfactory yields.Good to excellent enantioselectivities (80%-91%ee) could still be achieved when the electron-withdrawing substituents including F, Cl and Br are located at 6- or 7-position(entries 15–17 and 20–22).However, when the EWG are at 5-position and a CF3group at 7-position, moderate enantioselectivities (51%-70%ee) were obtained (entries 7–12 and 23).In the case of 4-substituted 3-diazooxindoles, 4-methyl failed to react with the cyclodiene possibly due to the steric hindrance at the 4-position.For the smaller 4-F substituent, the reaction could occur but besides prolonged time (24 h), a higher catalyst loading of 1.0 mol% and an elevated reaction temperature (25 °C) were also necessary because of the steric hindrance and the strong electronwithdrawing property of the fluorine (entry 3).The desired product 3b was generated in 62% yield with 78%ee.In addition, the effect of theN-substituent was studied as well.The reactions of unsubstituted and 5-Me substitutedN-benzyl diazooxindoles proceeded efficiently to afford the products 3a′and 3c′in 88% yield with 97%eeand 88% yield with 99%ee, respectively (entries 2 and 5).The results were slightly better in terms of both the yields and enantioselectivities when compared with the correspondingN-methyl reactants.On the other hand, the reaction of the 5-Cl substitutedN-benzyl diazooxindole provided an obviously inferior result to that of the 5-ClN-methyl diazooxindole (entry 9vs.8).The absolute configurations of the products 3c′(CCDC:1476610)and 3o (CCDC:2051865) were determined by X-ray single crystal diffraction analysis to beS(Fig.3).
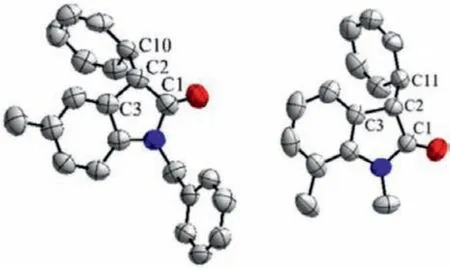
Fig.3.Molecular structures of compounds 3c′(left) and 3o (right).
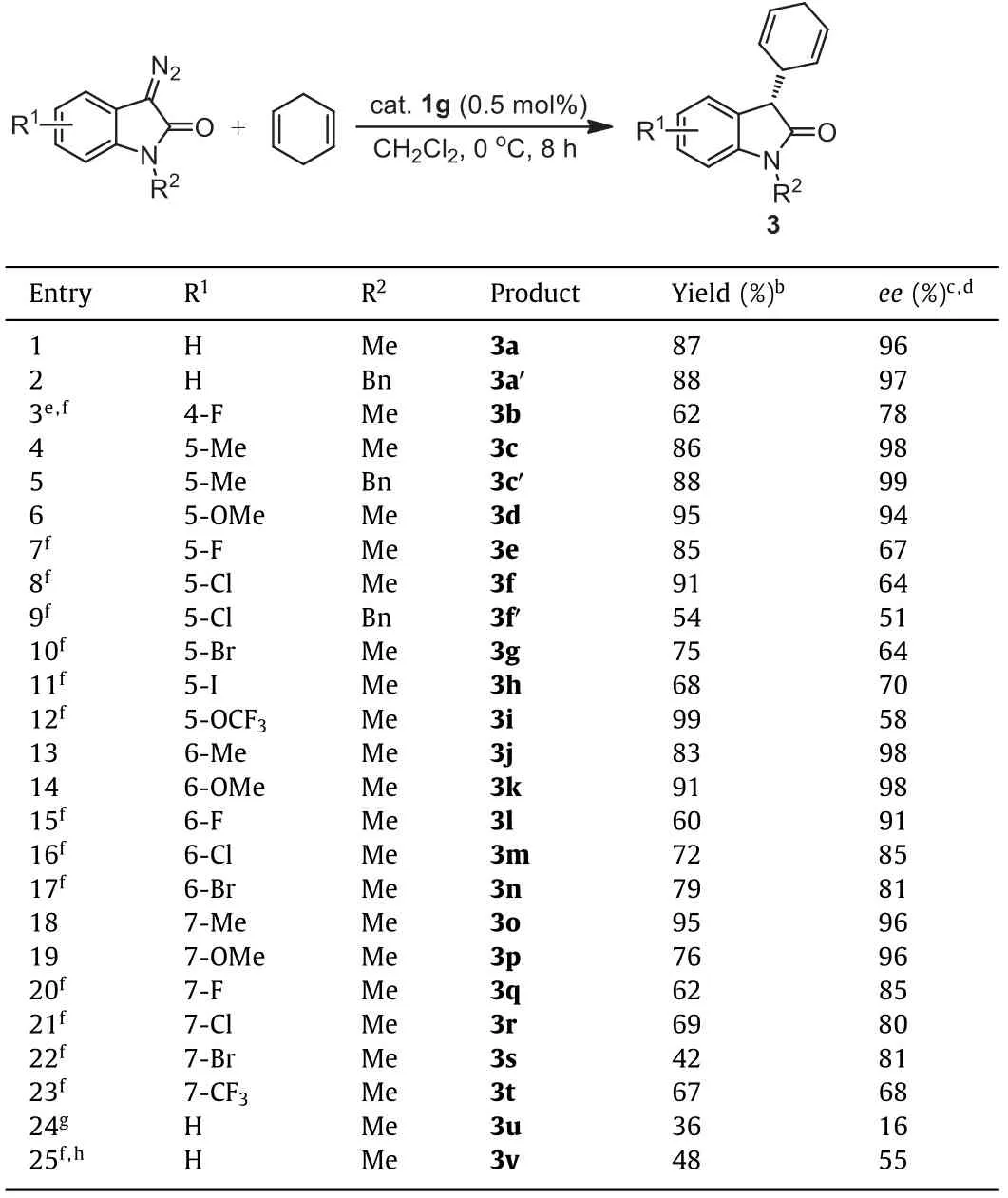
Table 2 Substrate scope.a
Besides 1,4-cyclohexadiene, 1-methyl-1,4-cyclohexadiene was also subjected to the reaction withN-methyl 3-diazooxindole,providing (S)-1-methyl-3-(m-tolyl)indolin-2-one 3u as the single regioisomer in 36% yield with 16%eeafter subsequent oxidation of the crude insertion product with DDQ.When 1,3,5-cycloheptatriene reacted with the same 3-diazooxindole, the corresponding insertion product 3v was obtained in 48% yield with 55%ee.In addition, we tried the C–H insertion reactions ofN-methyl 3-diazooxindole into cyclohexene and toluene.Unfortunately, no desired products were observed.
A scale-up reaction of 1-benzyl-3-diazoindolin-2-one with 1,4-cyclohexadiene was carried out on a 2 mmol scale under the optimized reaction conditions with a reaction time of 24 h (Scheme 1).The desired product 3a′was isolated in excellent yield without any loss in enantioselectivity (93% yield, 98%ee).In addition, the chiral cyclohexa-2,5–dien-1-yl group in the product 3a′was easily reduced by catalytic hydrogenation in the presence of Pd/C catalyst,delivering the corresponding 3-cyclohexyl substituted oxindole 4a′in 70% yield with 95%ee.
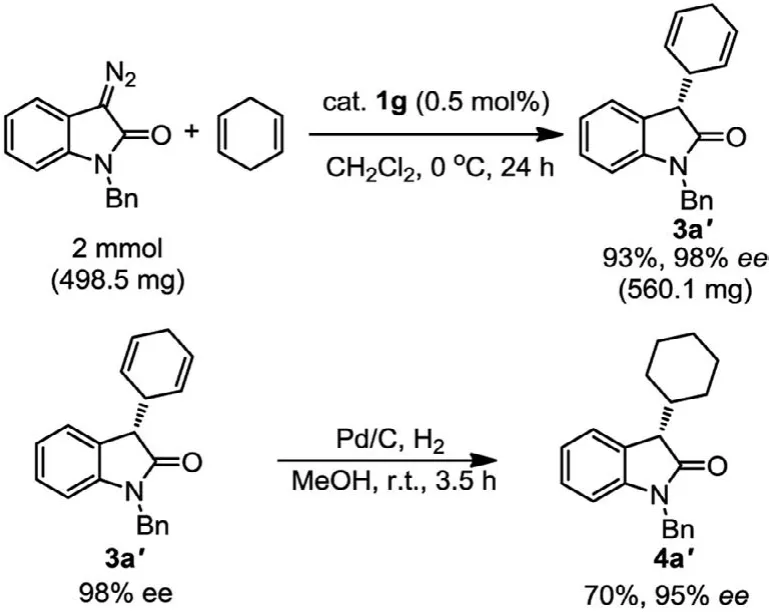
Scheme 1.A scale-up experiment and transformation of the catalysis product 3a′.
In order to gain a better understanding of the reaction mechanism and the origin of stereocontrol of the current pincer Ircatalyzed asymmetric C–H insertion, density functional theory(DFT) calculations were performed by employment of Gaussian 09 program [30].The geometry optimization was carried out by using the M06–2X [31,32] functional with the basis set of 6–31G(d,p) for C, N, Cl, O, H and SDD for Ir [33,34] and accounting for the dichloromethane solvent effect by employing the IEF-PCM[35,36] solvation model (M06–2X/6–31G(d,p)+SDD//IEF-PCMDCM).As shown in Fig.4, the reaction initiates with dissociation of the water from the catalyst 1g, which requires only 1.4 kcal/mol energy, giving rise to the formation of the 16-electron intermediatetrans-5 with the two chloride ligands locatedtransto each other.Then the resultingtrans-5 intermediate would interact with the 3-diazooxindole to generate the iridium carbenoid complex along with extrusion of nitrogen.The energy barrier associated with this process (through the transition state 8-TS) is found to be 25.9 kcal/mol, which is difficult to attain under the optimized reaction conditions.Meanwhile,trans-5 can rearrange to itsciscounterpartcis-5 through 6-TS with a much lower energy barrier of 17.5 kcal/mol.Furthermore, the interaction ofcis-5 intermediate with the 3-diazooxindole proceeds very readilyviathe 7-TS because the 7-TS has a lower energy thancis-5, furnishing the iridium carbenoid intermediate 9 with the carbene ligand coordinated at the axial position.Subsequently, the carbenoid intermediate 9 undergoes the C–H insertion step with 1,4-cyclohexadiene to give the product 3a.It was found that the reaction did not proceedviaa three-centered transition state which would yield directly the insertion product 3a.Instead, a stepwise process involving hydride transfer followed by C–C bond formation was proposed computationally (Fig.4).This is quite different from the related Ir and Rh catalyzed asymmetric C–H insertion with cyclohexadiene,where a concerted or concerted asynchronous mechanism involving a three-centered transition state was well supported by computational analysis [37,38].In the hydride transfer step,Si-face approach of the cyclodiene to the intermediate 9 through the 10-TS has an energy barrier of 15.3 kcal/mol, whereas a higher barrier of 20.2 kcal/mol (11-TS) is present for the correspondingReface approach.The higher energy of the 11-TS is related to the steric hindrance of upward 4S-phenyl substituent on the imidazoline ligand, which shields theRe-face of the carbenoid intermediate, thereby making theSi-face approach of the cyclodiene preferentially and leading to the formation of (S)-isomer of the product with high enantioselectivity.In addition, noncovalent interaction (NCI) [39,40] was used to analyze the transition states 10-TS and 11-TS (Fig.5).It is found that there are five stronger interactions (C–H···Cl, C–H···π, C–H···Cl and twoπ···π) in 10-TS.While only four interactions (C–H···π, C–H···Cl, N–H···πandπ···π) exist in 11-TS.The observation also indicates that 10-TS is more stable than 11-TS.The lower energy 10-TS results in generation of the zwitterionic intermediate 12.From this intermediate, the reaction continues and the C–C bond is formed to afford the product (S)-3aviathe 14-TS with an energy barrier of 10.7 kcal/mol.
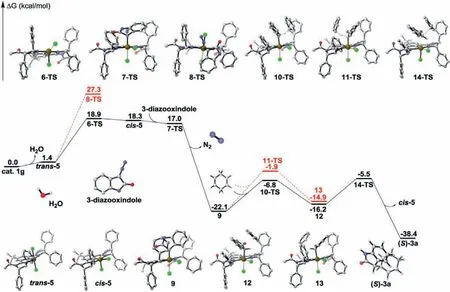
Fig.4.The relative Gibbs free energy profiles of the pincer (Phebim)Ir complex 1g catalyzed asymmetric C–H insertion of 3-diazo-1-methylindolin-2-one with 1,4-cyclohexadiene (light gray, white, blue, red, green and brown balls represent C, H, N, O, Cl, Ir atoms respectively).
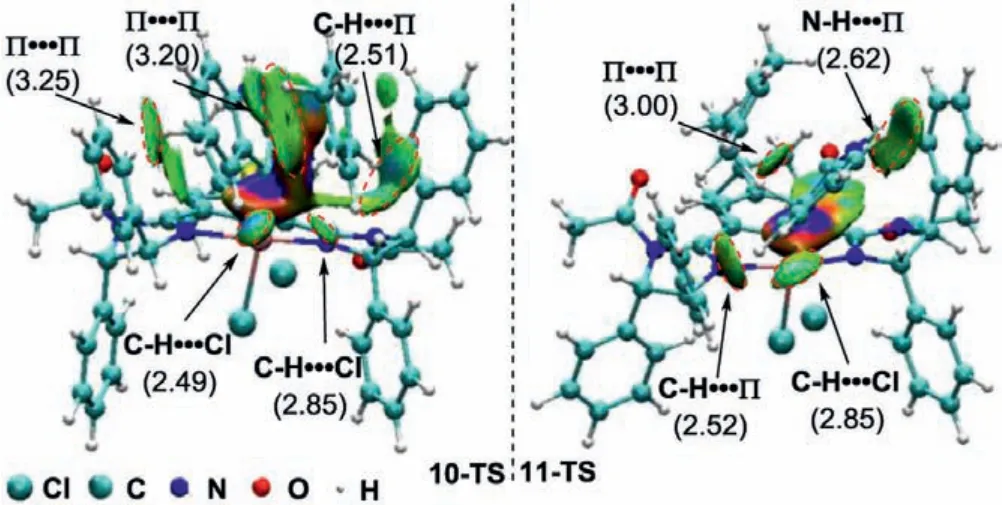
Fig.5.NCI analysis of the transition states 10-TS and 11-TS (distances in Å).
In summary, we have developed a chiral NCN pincer (Phebim)Ir complex-catalyzed enantioselective C–H insertion of 3-diazooxindoles with 1,4-cyclohexadiene.During the investigations four new (Phebim)Ir complexes were synthesized and well characterized.The catalytic reactions tolerate a variety of functional groups, furnishing chiral 3-substituted oxindoles with structural diversity in high yields and in most cases with good to excellent enantioselectivities (15 of 23 examples, 80%-99%ee).DFT calculations suggest that the current asymmetric C–H insertion proceedsviaa stepwise mechanism involving hydride transfer and the subsequent C–C bond formation, rather than a concerted or concerted asynchronous mechanism involving a three-centered transition state.The calculations also explain the stereoselectivity and are consistent with the experimental observation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
This research was supported by a grant from the National Natural Science Foundation of China (No.21472176).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.067.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction