
Chemo-, site-selective reduction of nitroarenes under blue-light,catalyst-free conditions
2022-06-20 07:59BinWangJiaweiMaHongyuanRenShuoLuJingkaiXuYongLiangChangshengLuHongYan
Chinese Chemical Letters 2022年5期
Bin Wang, Jiawei Ma, Hongyuan Ren, Shuo Lu, Jingkai Xu, Yong Liang, Changsheng Lu,Hong Yan
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering,Nanjing University, Nanjing 210023, China
Keywords:Nitroarenes Selective Reduction Catalyst-free Photoinduced Transborylation
ABSTRACT The tandem reaction of photoinduced double hydrogen-atom transfer and deoxygenative transborylation for chemo- and site-selective reduction of nitroarenes into aryl amines under catalyst-free, room temperature conditions was disclosed in excellent yields.In this reaction, isopropanol (iPrOH) was used as hydrogen donor and tetrahydroxydiboron [B2(OH)4] as deoxygenative reagent with green, cheap, and commercially available credentials.In particular, a wide range of reducible functional groups such as halogen(-Cl, -Br and even -I), alkenyl, alkynyl, aldehyde, ketone, carboxyl, and cyano are all tolerated.Moreover,the reaction preferentially reduces the nitro group at the electron-deficient site over another nitro group in the same molecule.A detailed mechanistic investigation in combination of experiments and theoretical calculations gave a reasonable explanation for the reaction pathway.
The prevalence of functionalized anilines in dyes, agrochemicals, polymers and biologically active compounds, has inspired the discovery of new patterns to reduce nitroarenes chemoselectively[1–5].Furthermore, the chemo-, site-selectivity in reduction of a particular nitro motif over other reducible functionalities or another nitro group in the same molecule is critically important for further derivatization, as to avoid the stepwise processes and the production of wastes [6,7].Traditional methods of reducing functionalized nitroarenes resulted in severe environmental issues by using an excess of metal reductants, such as Zn, Fe, Sn, combined with acids [8–12].To solve this problem, several groups [1,5,13–21] have examined the reactivity and selectivity in converting the functionalized nitroarenes to aryl amines by using modified and tailor-made metal nanoparticles or other solid catalysts.Additionally, the requirement of gaseous H2, high temperature and pressure in these catalytic methods, may cause potential problems [22–25].
Evidently, from a sustainable perspective, the photo-driven process is an attractive strategy for the preparation of functionalized anilines due to easy-handling, environmentally-benign and green credentials [26–31].To date, photo-induced protocols, using semiconductor [27,29], plasma [32], dye-sensitizer [27,33] and photocatalysts [33,34], promoted the transformation of aromatic nitro compounds to the corresponding aryl amines, however some side reactions were involved in these processes [35].Interestingly,the process of direct hydrogen abstraction by photo-excited nitroarenes fromiPrOH [36] or hydrazine [37] is feasible under catalyst-free conditions, which demonstrated a simple means to activate functionalized nitro compounds.However, this protocol merely provided the conversion of nitroarenes to aryl hydroxylamines and/or a mixture of azoxy-, azo-compounds.Therefore,the practical photo-driven patterns without catalysts for chemoselective reduction of highly functionalized nitro- to NH2-bearing molecules remain to be explored.
In this context, inspired by the oxophilic nature of diboron reagents (e.g., B2pin2, B2nep2, B2(OH)4) which could be used as efficient deoxygenative additives for the reduction of nitro, amineNoxides and carbonyl compounds with their commercially available and green characteristics [38–47], we have developed a tandem protocol of catalyst-free [48], photo-induced double hydrogenatom transfer (HAT) [49] and transborylative deoxygenation for the selective reduction of nitro motifs into NH2groups with bluelight irradiation at room temperature (Scheme 1).The protocol employediPrOH [50] and B2(OH)4[51] as reductants with their cheap and eco-benign advantages.Of note is this protocol highlights the excellent site selectivityviaa double HAT fromiPrOH to a photoexcited certain nitro group, avoiding the hydrogenation of other reducible sites.
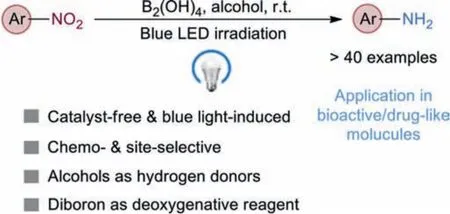
Scheme 1.Present strategy for nitro reduction under blue light irradiation.
Initially, 4-nitroacetophenone (1a) was selected as a model substrate to optimize conditions for the selective reduction of nitro compounds.Our investigation began with searching for a hydrogen donor alcohol by treating 1a with alcohols, B2pin2in DCE solvent with the blue LED (400 nm) irradiation at room temperature.The reductive process occurred smoothly in a good yield with an excellent selectivity.By selecting isopropanol as hydrogen donor, product 2a was detected as the sole product in 89% yield by1H NMR(Table 1, entry 4).Methanol or ethanol gave slightly lower yields in 62% and 76%, respectively (Table 1, entries 1 and 2).However,the use of HFIP (hexafluoroisopropanol) afforded 2a in only 29%yield (Table 1, entry 3).The transformation of nitro group did not occur in dark (Table 1, entry 7), and only trace amount of 2a or no product could be generated without either B2pin2or isopropanol(Table 1, entries 6 and 5).Both bis(neopentylglycolato)diboron(B2nep2) and B2(OH)4were suitable for our system, delivering 2a in 87% and 90% yield, respectively (Table 1, entries 8 and 10), but bis(catecholato)diboron (B2cat2) showed a relatively lower yield of 62% (Table 1, entry 9).Notably, the yield of 2a can be improved to 85% or more than 95% by using toluene or THF as the solvent(Table 1, entries 13 and 14).To our delight, protic solvents, such as isopropanol and ethanol, also gave rise to excellent yields (Table 1,entries 11 and 12).The reaction also occurred when ethanol was used as both solvent and hydrogen donor in the absence of isopropanol, but led to slightly lower yield (Table 1, entry 16).Taking the cost and environment-friendly features into considerations, the optimal reaction conditions are to employ B2(OH)4as the deoxygenative reagent, ethanol as the solvent, unless otherwise specified.
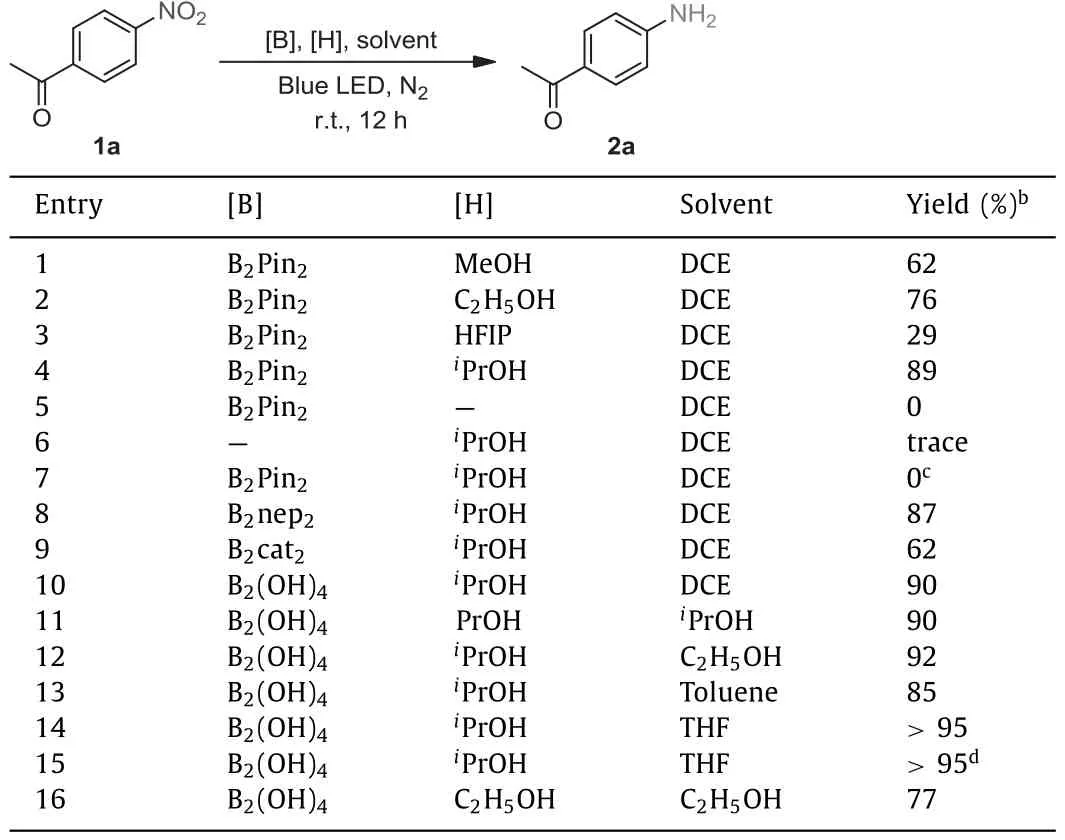
Table 1 Reaction optimization.a
With an optimized condition in hand, the scope of nitro substrates was extensively investigated (Scheme 2).First, the chemoand site-selectivity was evaluated for a wide range of nitroarenes bearing the reducible groups, including ketone, aldehyde, cyano,halide and ester moieties.Remarkable functional group tolerance was demonstrated, affording the desired products in good to excellent yields (2a-2e, 2g, 2k-2m, 2r, 2s, 2u).It is noteworthy that the terminal alkynyl, even terminal alkenyl groups were also tolerated under standard conditions (2h, 2v).Moreover, the process is site-selective for nitroarenes with two nitro groups in the same molecule.For example, theortho-nitro of fluorinated dinitrobenzene was selectively reduced with thepara-nitro motif almost unscathed (2i); the nitro-site reduction was preferred at an electrondeficient aryl moiety in a dinitro-substituted diaryl molecule (2j,2x); the nitroethyl-substituted nitrobenzene was smoothly reduced in 77% yield, with the nitro group on the alkyl moiety remaining intact (2w).Next, the catalyst-free reduction protocol was employed successfully in theortho-, meta-, andpara-substituted substrates, as shown in Scheme 2, generally providing an excellent level of yields, in contrast to the reported methods which were prone to dehalogenation forortho-, meta-substituted halonitrobenzenes [1,2].It was observed that the stronger the electronwithdrawing ability of the substrates is, the higher the reaction rate is.We obtained electron-rich aromatic amines in moderate to good yields by changing solvent and prolonging reaction time.To our surprise,ortho-dimethylnitrobenzene shows the reactivity similar to those of the electron-deficient substrates.Several heterocyclic compounds containing nitro groups are also proven to be amenable (2x, 2ac-2ak).However, the aliphatic nitro compounds could only afford the corresponding amines (i.e.2al) in low yield.To further demonstrate the potential pharmaceutical application,several marketed nitro substituted drugs were tested, delivering amino-containing products in moderate to excellent yields (2am-2aq).Finally, we performed several gram-scale reactions for selected substrates.In these cases, similar yields to the 0.2 mmol scale transformations were obtained (2c, 2g, 2ab).
To propose a plausible mechanism, we first conducted the reaction of 1a with 15 equiv.of isopropanol in THF with blue LED irradiation under N2atmosphere for 5 h without the addition of diboron reagent, which resulted in hydroxylamine 4a in 95%yield together with the formation of acetone as detected by GCMS (Scheme 3a and Fig.S3 in Supporting information).Meanwhile,the addition of 2.0 equiv.of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) led to the dramatically decreased yield of 4a along with the observation of TEMPO-H by HRMS analysis (Scheme 3b and Fig.S2 in Supporting information).This suggests that an electron-deficient nitroarene can be directly reduced toN-aryl hydroxylamine by only using isopropanolviaa photoinduced radical hydrogen transfer process.Next, when replacing 4-nitroacetophenone with nitrobenzene, and prolonging the reaction time to 12 h, a moderate yield of azoxybenzene along with a trace amount of nitrosobenzene was obtained (Scheme 3c), suggesting that nitrosoarene is an intermediate between nitroarene andN-aryl hydroxylamine.For a less electron-deficient substrate, the photoinduced reduction of nitrosoarene by isopropanol slows down to allow formation of azoxybenzene byproduct.However, the treatment of nitrosobenzene with 1.5 equiv.of B2nep2afforded a high yield of diboryl intermediate 5q within 40 min.The hydrolysis of intermediate 5q proceeded quickly at 0 °C, delivering hydroxylamine 4q in 82% yield (Scheme 3d).Therefore, we concluded that isopropanol could reduce nitroarenes to nitrosoarenes, and diboron is efficient to reduce nitrosoarenes toN-aryl hydroxylamines.Finally, to our surprise, the hydroxyaniline underwent a fast transborylative deoxygenation within 5 min in the presence of only 1 equiv.of B2nep2, giving the aniline product in almost quantitative yield (Scheme 3e), and the generated O(Bnep)2was detected by1H NMR,11B NMR and GC-MS (Figs.S9–S11 in Supporting information).
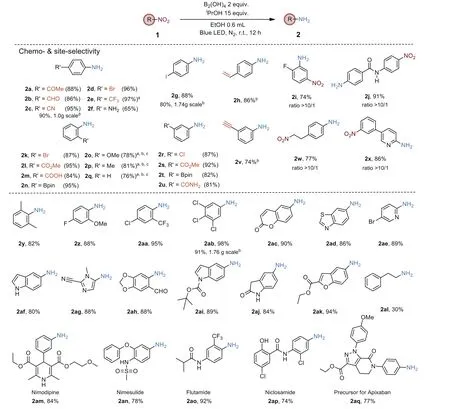
Scheme 2.Substrate scope of nitro compounds.General conditions:substrate (0.2 mmol), tetrahydroxydiborane (0.4 mmol), isopropanol (15 equiv.), solvent (0.4 mL), N2,at room temperature for 12 h, with 400 nm LED irradiation.Isolated yields unless otherwise specified.a NMR yield.b 24 h.c The solvent was replaced by THF with the addition of 5 equiv.of H2O.
To rationalize the overall reaction and to gain mechanistic insights, we conducted DFT calculations (B3LYP-D3 with solvation[52–56]).As shown in Scheme 4, ground-state nitrobenzene (R’)is photoexcited to triplet nitrobenzene (R), which has a diradical character.R abstracts a hydrogen atom from the secondary carbon of isopropanolviaTS1, which has a barrier of only 10.2 kcal/mol,forming two radicals (INT1).Then a faster second HAT processviaTS2 liberates acetone and givesN-hydroxy-N-phenylhydroxylamine(INT2), which continues to dehydrate to yield nitrosobenzene(INT3).The reaction between nitrosobenzene and diboron was previously studied by DFT calculations and determined as a concerted mechanism [57].We also found a single transition state TS3 connecting INT3 and INT4 with a barrier of 15.6 kcal/mol.With a stoichiometric amount of water (previously released by INT2) in the system, INT4 can quickly hydrolyze to yieldN-phenyl hydroxylamine (INT5).
We then discovered a unique mechanism for the final reductive stage, fromN-phenyl hydroxylamine to aniline.We supposed that an anionic oxygen, compared to the neutral oxygen in hydroxyl,could have a better affinity with diboron.As the generation of zwitterionic form of hydroxylamine is slightly endergonic from its neutral form [58], aniline oxide (INT6) could be the actual reactive species.In our calculations,N-phenyl hydroxylamine firstly transforms to INT6viaproton transfer.Then INT6 binds to diboron to form INT7.Finally, the non-bonded boryl group migrates to oxygen and simultaneously breaks the N-O bondviatransition state TS4.As a result, the overall barrier of this stage is 19.0 kcal/mol, which is consistent with our experimental observation of a fast reaction at room temperature.A concerted pathway in whichN-phenyl hydroxylamine remains neutral form (INT5-TS4’-P) was also located with a barrier as high as 30.2 kcal/mol, which is not plausible for this reaction.
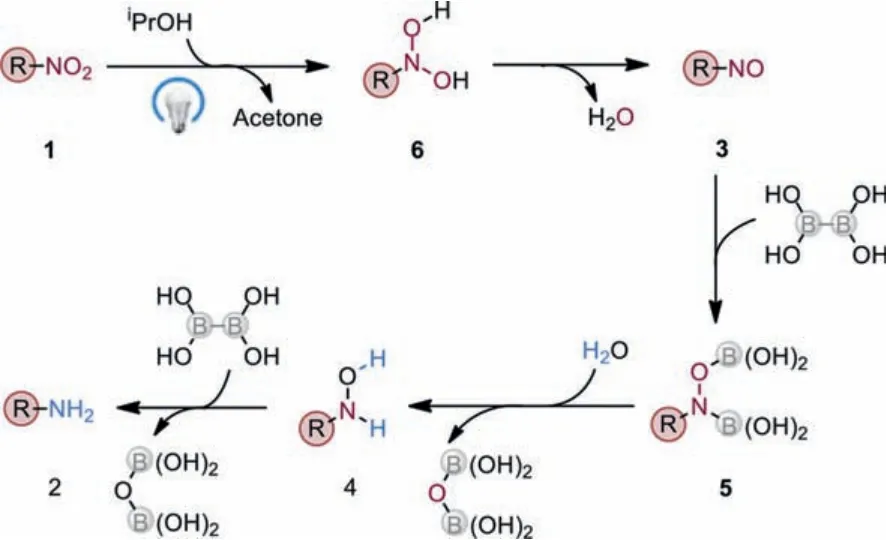
Based on our controlled experiments and the DFT calculations,we proposed a reaction mechanism as shown in Scheme 5.Initially, double hydrogen atom transfer between photoexcited nitro compound 1 and isopropanol gives dihydroxylamine 6.Then intermediate 6 is transformed to a nitroso-intermediate 3 with the loss of one water molecule.Subsequently, a fast diborylation of 3 with a diboron reagent delivers aN,O-diboryl intermediate 5,which quickly reacts with water to provide hydroxylamine 4.Finally, the transborylative deoxygenation of 4 gives the final product 2.
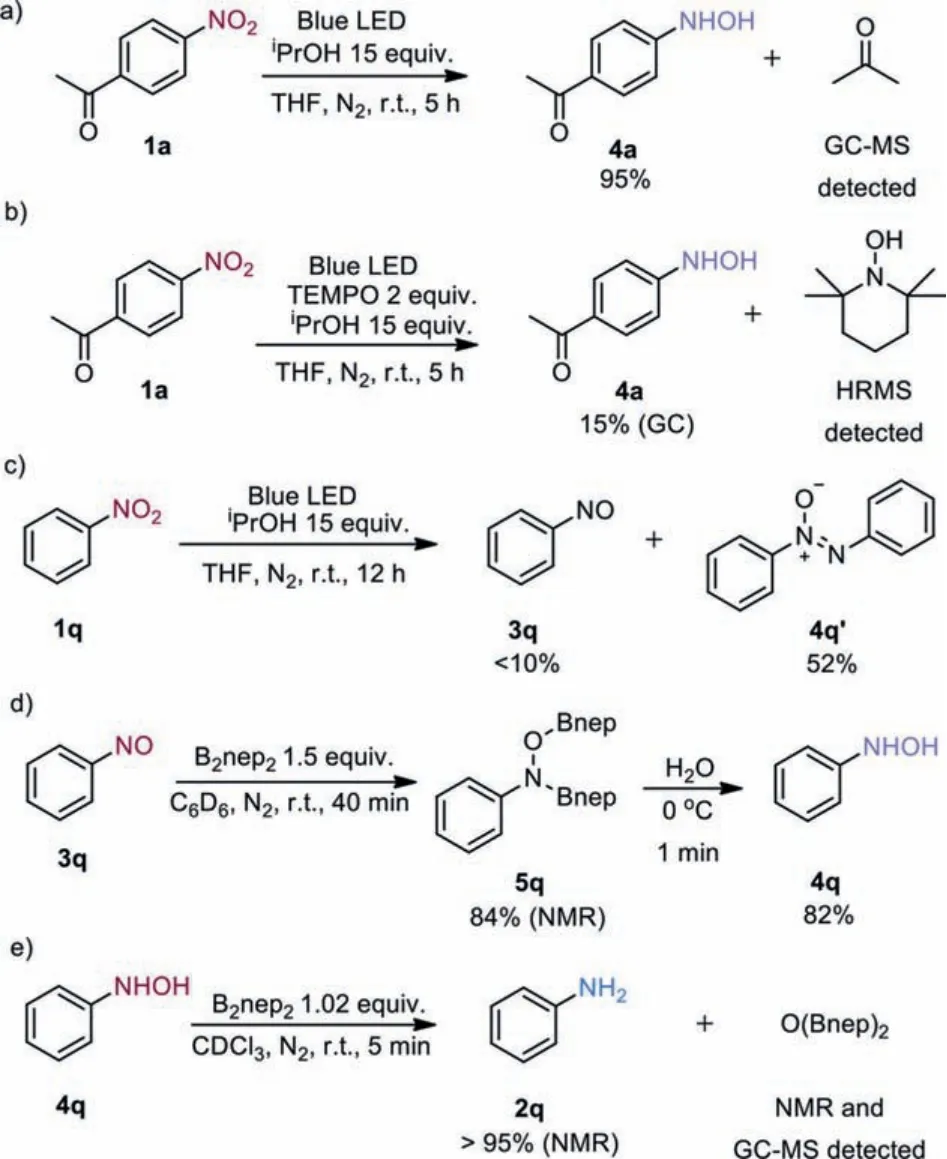
Scheme 3.Probing mechanistic studies.

Scheme 4.Probing energy profile of the proposed reaction mechanism.Values are given as relative Gibbs free energy at 298 K, 1 atm, in kcal/mol, computed with B3LYPD3(BJ)/6–311+G(d,p) (SMD, iPrOH)//B3LYP-D3(BJ)/6–31G(d) (SMD, iPrOH).a In triplet state.b In singlet state.
Scheme 5.Proposed mechanism.
In summary, a catalyst-free, photo-induced method for the chemo- and site-selective reduction of substituted nitroarenes into primary amines has been developed.This protocol employsiPrOH as a hydrogen donor, and B2(OH)4as a transborylative deoxygenation reagent with green, cheap, and commercially available advantages.It tolerates a wide range of reducible functional groups, such as halogen (-Cl, -Br and even -I), alkenyl, alkynyl, aldehyde, ketone, carboxyl and cyano.Meanwhile, our protocol preferentially reduces the nitro group at the electron-deficient site over another nitro group in the same molecule.A detailed mechanistic investigation through both experiments and theoretical calculations gave a reasonable explanation for the reaction pathway.Additionally, we used this method for the selective reduction of highly functionalized pharmaceutical molecules and the gram-scale syntheses.This protocol will be potentially useful in future applications.
Declaration of competing interest
The authors report no declarations of interest.
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (Nos.21820102004, 91961104,21803030, 21875104, and 51673095), Program B for outstanding Ph.D.candidates of Nanjing University, the Fundamental Research Funds for the Central Universities (No.020514380253), the Natural Science Foundation of Jiangsu Province (No.BK20211555), and the Jiangsu Innovation and Entrepreneurship Talents Plan.We thank the High Performance Computing Center (HPCC) of Nanjing University for doing the numerical calculations in this paper on its blade cluster system.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.023.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction