
Dehydrative Beckmann rearrangement and the following cascade reactions
2022-06-20 07:59YongjioWeiYinghuiLiuLnGuiXie
Chinese Chemical Letters 2022年5期
Yongjio Wei, Yinghui Liu, Ln-Gui Xie,*
a School of Chemistry and Materials Science, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, Nanjing Normal University, Nanjing 210023, China
b College of Life Sciences, Nanjing Normal University, Nanjing 210023, China
Keywords:Beckmann rearrangement Multiple components reaction Amide Thioamide Tetrazole
ABSTRACT The Beckmann rearrangement has been predominantly studied for the synthesis of amide and lactam.By strategically using the in situ generated Appel’s salt or Mitsunobu’s zwitterionic adduct as the dehydrating agent, a series of Beckmann rearrangement and following cascade reactions have been developed herein.The protocol allows the conversion of various ketoximes into amide, thioamide, tetrazole and imide products in modular procedures.The generality and tolerance of functionalities of this method have been demonstrated.
Beckmann rearrangement has been well established as an alternative route to the synthesis of amide and lactam.Its widely applications across synthetic laboratory and industry are represented by the synthesis of paracetamol from the oxime derivative of 4-hydroxyacetophenone and the process of producing caprolactam from cyclohexanone oxime, which is used as the monomer of nylon-6 [1–3].The classical condition of Beckmann rearrangement requires the use of strong acid and high temperature conditions, which limits the application of this transformation.Consequently, in the past decades, much interest has been attracted from the organic chemists by the development of catalytical procedures, to force the conversion of oximes to amides [4].Therefore, many elegant protocols have been presented, with the effort to use organo-molecule and Lewis acid as the catalyst or cocatalyst [5–15].Recently, photo-induced procedures were developed to facilitate the rearrangement as well [16–18].However,Beckmann rearrangement is predominantly employed for the construction of synthetic architectures with amide or lactam unit, because the leaving -OH group from the oxime is in the system and inevitably plays the role of nucleophile attacking to the nitrilium intermediate to form the amide precursor (Scheme 1A), though special oximes with intramolecular nucleophilic atoms have been witnessed cyclization [19].While the addition of intermolecular nucleophiles to the nitrilium intermediate, in successful examples,requires the pre-conversion of OH on the oxime to the corresponding OTs or OMs, which are of much weaker nucleophilicity [19].
Ugi type multiple components reactions (MCRs) are of the utmost importance in the construction of molecular diversity in research and development in the pharmaceutical companies [20,21].Mechanistically, Ugi type MCRs are cascade reactions starting with the generation of iminiums from amines and aldehydes in the presence of carboxylic acids, following by the addition of isocyanides to the iminiums, deliveringα-amino nitrilium intermediates (Scheme 1B), which are the well accepted key intermediate of this transformation and eventually producing the correspondingα-amino products, such as amides and tetrazoles, in the presence of different nucleophiles [22,23].We speculate that upon the treatment of oxime with a dehydrating reagent [24–26], a nitrilium intermediate would be produced after the classic Beckmann rearrangement, and ready for attacking by a nucleophile.These whole transformations should be able to make the possibility of preparing diverse products (amides, tetrazoles, imides and so on) from oxime derivatives in a similar fashion of the Ugi type MCRs.And notably, the products would be structurally more general, because of the absence ofα-amino group comparing to the products of Ugi MCRs.
Appel’s salt, combination of triphenylphosphine (Ph3P) and carbon tetrachloride (CCl4), is often used as dehydrating agent [27–29].It is known that catalytic amount of Appel’s salt can be used to capture the OH from oxime and thus promotes its selfpropagating process at elevated temperature [30].We hypothesize that a stoichiometric Appel’s salt would possibly be able to remove the OH group by forming a highly stable P-O double bond [19],and would then allow nitrilium ion intermediate to accept intermolecular nucleophiles, delivering various products with the similar mechanism of Ugi type reactions (Scheme 1C).
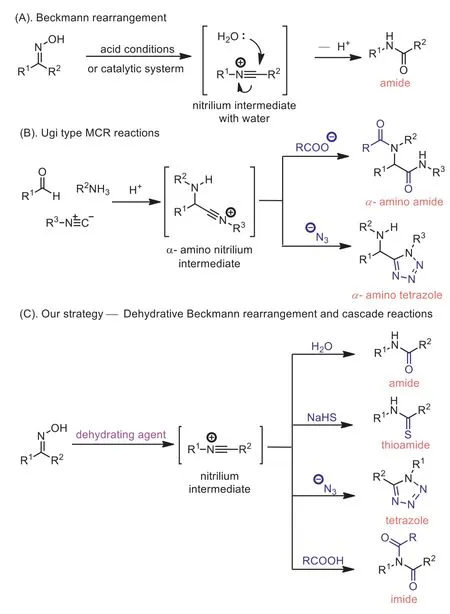
Scheme 1.(A) Classical Beckmann rearrangement.(B) Ugi type multiple components reactions (MCRs).(C) Dehydrative Beckmann rearrangement and cascade reactions.
The concept was initially tested by submitting phenylbutanone derived ketoxime to the conditions of varying amounts of triphenylphosphine (Ph3P) and tetrachloride carbon (CCl4) with various solvents, such as dichloromethane (DCM), acetonitrile and dichloroethane (DCE), which after hydrolysis delivered the target amide 1 in moderate yields generally (Table S1 in Supporting information for details).After optimization, the conditions of 2 equiv.of Ph3P and 1.2 equiv.of CCl4inN,N-dimethylformamide (DMF 0.1 mol/L) were found suitable to produce the amide 1 in 80% yield at ambient temperature (Scheme 2).Reducing the amount of Ph3P to 1.5 equiv.led to relatively lower yield (66%).
We then tested the scope of the oxime with the optimized conditions.Represented examples were shown in Scheme 2.Excellent yield of amide 2 was obtained after the rearrangement of the oxime derived from naphthalenylethanone.Electron-rich phenyl rings (3 and 4) could be introduced to the amides by applying the corresponding oximes to the conditions.Halide functionalities represented by bromide, trifluoromethyl and trifluoromethoxyl groups on the aromatic were proved deliverable (5–7).Heteroarene, thiophene was applicable to this procedure (8).Vinyl substituted acetaniline (9) andN-vinyl acetamide (10) were successfully prepared in reasonable yields.A cyclopropane derived amide 11 could be synthesized smoothly.In addition, oximes deriving from dibenzyl ketone and macroketone were amenable to the rearrangement as well, delivering the corresponding amide and macrolactam in synthetic useful yields (12 and 13).Furthermore,18O labelled amide 14 were proved accessible by the use of H218O in the hydrolysis stage in this amide synthetic sequence.
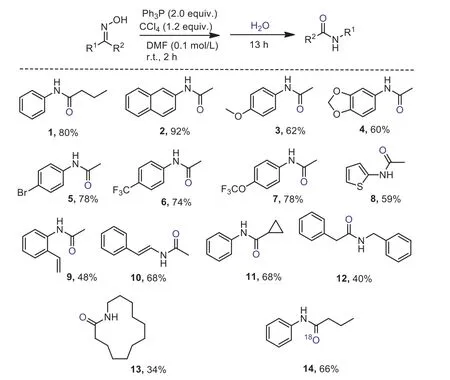
Scheme 2.Scope respect to the synthesis of amide.Standard condition:ketoxime 0.30 mmol, Ph3P 0.60 mmol, CCl4 0.36 mmol, DMF 3.0 mL; isolated yields are given.
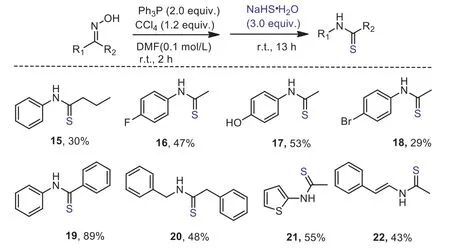
Scheme 3.Representative synthesis of thioamide.Standard condition:ketoxime 0.30 mmol, Ph3P 0.60 mmol, CCl4 0.36 mmol, NaHS·H2O 0.90 mmol, DMF 3.0 mL;isolated yields are given.
Thioamide motif is important structural unit, that has been extensively studied in the synthesis of heterocycles and amino molecules [31,32].Modified peptides with one or more thioamide in the backbones have been found increased pharmacokinetic activity [33] or enhanced half-life [34,35] in the studies of drug discovery.Prevalent methods for the synthesis of thioamides are related to the use of phosphorus pentasulfide or Lawesson type reagents to converse the corresponding carbonyl analogues [36,37].Though Bachmann rearrangement has been vastly explored for the synthesis of amide, directly transferring ketoxime to thioamide is rare and rather limited to the use of P=S type thiolation reagent[38–40].As part of our dehydrative strategy on the Beckmann rearrangement, we applied sodium hydrosulfide in the stage of hydrolysis, and were delighted to find the corresponding thioamide 15 was detected and separatable (Scheme 3).A brief test showed that ketoximes derived from substituted aryl alkyl ketones were amenable to the new synthetic procedure for thioamide (15–18).Diaryl thioamide 19 was obtained in excellent yield by submitting the corresponding ketoxime to the standard conditions.Dibenzyl,heteroaryl/alkyl andN-vinyl thioamides (20–22) were suitable targets for this synthetic method.
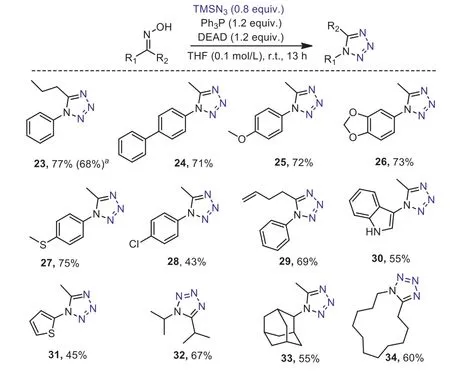
Scheme 4.Scope respect to the synthesis of tetrazole.Standard condition:ketoxime 0.30 mmol, Ph3P 0.36 mmol, DEAD 0.36 mmol, TMSN3 0.24 mmol, THF(tetrahydrofuran) 3.0 mL; isolated yields are given. a The reaction was carried out on 8 mmol scale.
The tetrazole [41,42] synthesis from phenylbutanone ketoxime[43,44] was initially conducted with Appel’s salts and trimethylsilyl azide, yielding the target product 23 in 39%, with corresponding amide as the major product.Optimization showed that employing Mitsunobu’s zwitterionic adduct [45–49], generatedin situby mixing Ph3P and diethyl azodicarboxylate (DEAD) in tetrahydrofuran,as the dehydrating agent was essential (Table S2 in Supporting information for details), leading to 77% isolated yield of the tetrazole 23 (Scheme 4).The use of chloride solvents (DCM, DCE) resulted in decline in yield.Aryl methyl ketoximes with electron-rich phenyl rings (24–27) or phenyl with slightly electro-withdrawing substitution such as chloride (28), underwent the tetrazole formation smoothly in the standard conditions.The success of introducing a terminal double bond into the tetrazole molecule (29), indicated the compacity of this transformation with the well-established various alkene derivations [50].Heteroaromatic rings, indole and thiophene substituted tetrazoles were obtained in synthetic useful yields (30 and 31), further highlighted the utility of this protocol.Synthesis ofN-aliphatic tetrazoles (32 and 33) were achievable by submitting the corresponding ketoximes to this procedure.In addition, cyclic ketoxime was also applicable to the conditions, affording the cyclic tetrazole 34 in 60% yield.Furthermore, carrying out the tetrazole synthesis in gram scale without further optimization of the conditions, afforded the product 23 in 68% yield.
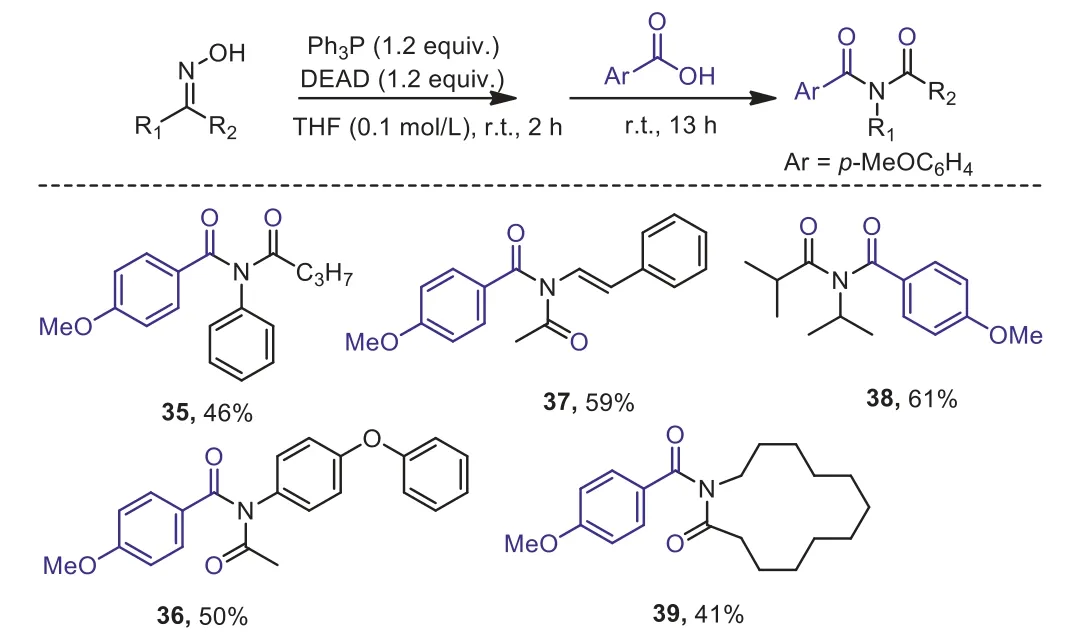
Scheme 5.Representative synthesis of imide.Standard condition:ketoxime 0.30 mmol, Ph3P 0.36 mmol, DEAD 0.36 mmol, para-anisic acid 0.45 mmol, THF 3.0 mL; isolated yields are given.
Furthermore, usingpara-anisic acid instead of azide source after the rearrangement of phenylbutanone oxime in the presence of Ph3P and DEAD, imide product 35 was isolated in moderate yield.Mechanistically, anisic acid played the role of a nucleophile in the addition to the nitrilium intermediate, which generated from the Beckmann rearrangement of the aryl alkyl ketoxime(Scheme 5).The formed adduct then underwent a Mumm rearrangement [51] to facilitate the formation of imide.More examples demonstrated that acyclic and cyclic dialkyl ketoximes were all able to be employed for the synthesis of corresponding imides in reasonable yields (36–39).
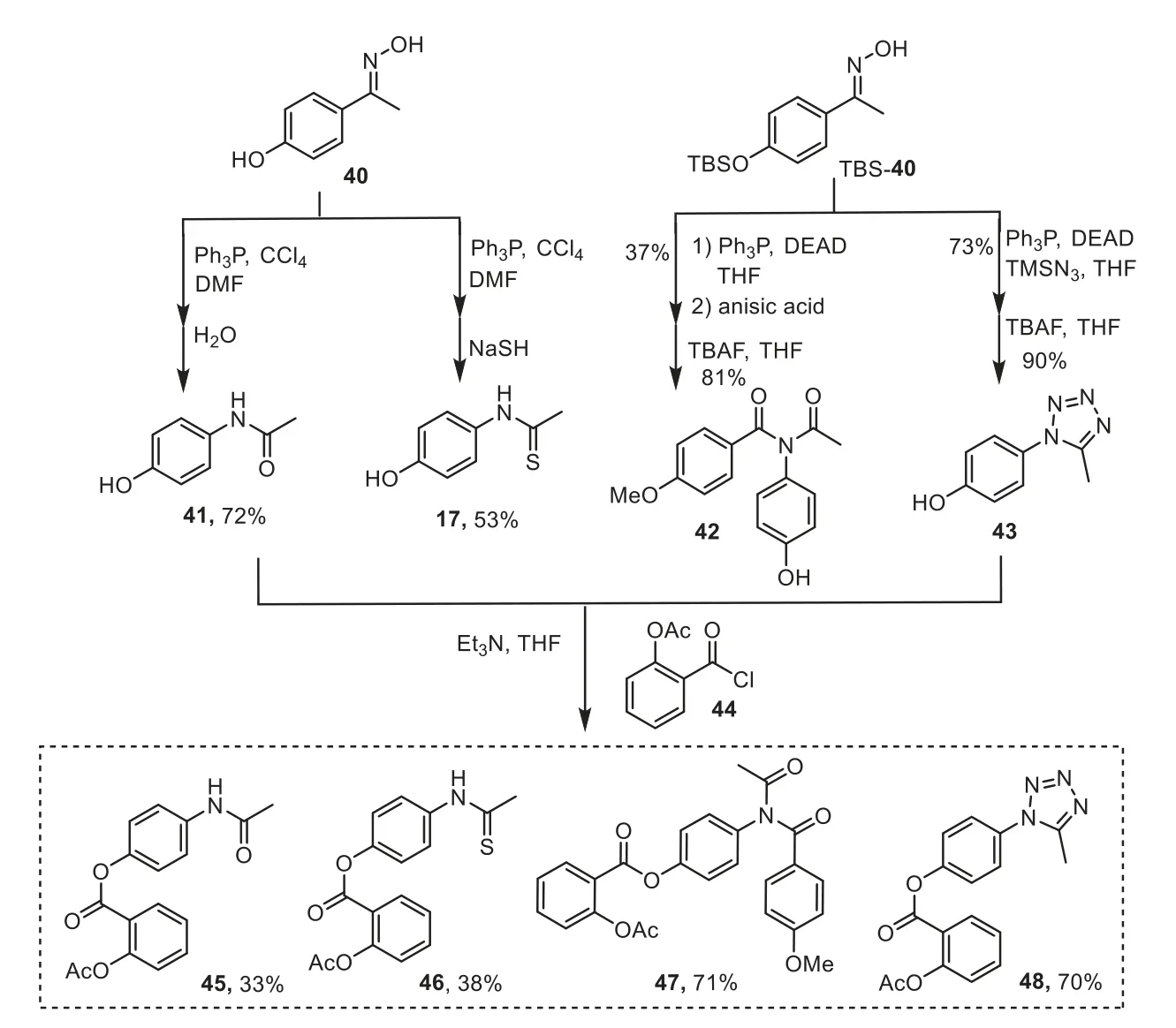
Scheme 6.Synthesis of Benorilate and its derivatives.
To showcase the utility of our dehydrative Beckmann rearrangement and following cascade reactions, we set to use the ketoxime 40 (Scheme 6) to undergo the rearrangement in the presence ofin situformed Appel’s salt, following by hydrolysis or sodium hydrosulfide addition, affording the amide or thioamide intermediator(41 and 17) respectively.While under the Mitsunobu’s zwitterionic adduct conditions,O-TBS protected 40 was applied for the synthesis of imide 42 and tetrazole 43 with an additional simple deprotection stage.The precursors were then esterified with acyl chloride 44 respectively, enabling the access to Benorilate (45) [52] and its thioamide, imide and tetrazole analogues (46–48).
In summary, by employing thein situgenerated Appel’s salt or Mitsunobu’s zwitterionic adduct as the dehydrating agent, Beckmann rearrangement and a series of following cascade reactions in the presence of different nucleophiles has been developed, facilitating the diverse synthesis of amide, thioamide, tetrazole and imide products from ketoxime in modular sequences.The wide scope of ketoximes and the tolerance of various functionalities in these reactions has been presented.The power in organic synthesis of these transformations have been evaluated by the modular synthesis of Benorilate and its derivatives.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by National Key Research and Development Project (No.2021YFC2100100), National Natural Science Foundation of China (No.21901123), Natural Science Foundation of Jiangsu Province (No.BK20190694) and Jiangsu Specially Appointed Professor Plan.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.10.020.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction