
Well-defined phosphate yttrium dialkyl complexes for catalytic stereo-controllable 1,4-polymerization of isoprene
2022-06-20 07:59PusuYngHuiZhenDuXingYuZhngYongLingXiJunToSunQinPengBingToGun
Chinese Chemical Letters 2022年5期
Pusu Yng, Hui-Zhen Du, Xing-Yu Zhng, Yong-Ling Xi, Jun-To Sun, Qin Peng,Bing-To Gun
a College of Chemistry, Nankai University, Tianjin 300071, China
b Department of Chemistry, Fudan University, Shanghai 200438, China
Keywords:Phosphate Yttrium alkyls Isoprene Stereo-controllable 1,4-Polymerization
ABSTRACT Oxygen ligation is envisioned to provide a stable and distinctive coordination environment to the strongly oxophilic rare-earth metals.However, the well-defined dialkyl complexes bearing oxyanion ancillary ligand had been rarely addressed for the instability of the complexes and the shortage of easily available ligands.Herein, we report the synthesis of phosphate ligated dialkyl yttrium complexes (PYR2) featuring a high stability and a tunable ligand.Treated with the borate reagent, the phosphate yttrium complex displays high activity and selectivity in the catalytic cis-1,4-polymerization of isoprene (up to 96.5%).Furthermore, using AlMe3 as an additive, the stereoselectivity switches to trans-1,4-polymerization (up to 92.0%).
Polyisoprene (PIP) with well-defined microstructure represents an essential class of functional material in elastomers and plastics [1–3].The huge demand of them thereby promotes an impressive development of rare-earth metal catalysts [4–6], which achieved the selective synthesis ofcis-1,4-PIP, [7–26]trans-1,4-PIP[27–33] and 3,4-PIP [34–39].It is noteworthy that some catalytic systems were reported to enable a switch of the selectivity by a slight modification of the metal center, ancillary ligand, or cocatalysts [40–54].For example, Anwander found that the lanthanum fluorenyl half-sandwich catalyst efficiently producedtrans-1,4-PIP(80%), while the lutetium complex gave access to highercis-1,4-contents (78.2%) [40].Arnold discovered that half-sandwich scandium borohydrides complexes with different NHC ligands could swtich the stereoselectivity in the isoprene polymerization (94%trans-1.4 and 80%cis-1.4) [41].Hou and Zhang reported that the yttrium amidinate complex, by addition of AlMe3, were transformed to heterotrinuclear Y/Al complex, which could dramatically switch the regio- and stereoselectivity of isoprene polymerization (from 3,4-isospecific to 1,4-cisselective) [46,47].In these cases, the precise control on the regio- and stereoselectivity definitely displays the state of the art in catalyst design.A tailor-made catalyst with new structure, good stability, high catalytic activity and selectivity, however, is still highly desired in both academic and industrial fields.
Dialkyl rare-earth complexes bearing a monoanionic ancillary ligand have recently drawn much attention, serving as efficient catalysts for the polymerization and functionalization reactions of alkenes [55–60].Most of these ligands feature delocalized carbanions or multidentate nitrogen anions structures (Scheme 1A).According to the strong oxophilicity of rare-earth metals, oxyanions are supposed to have great potential for acting as suited ancillary ligands [61–75].Based on this assumption, Evans synthesized dialkyl rare-earth complexes adopting steric bulky 2,6-di–tert-butylphenol as an ancillary ligand [61,62].Subsequently, functionalized phenols [63–67], silanols [68,69], alkoxy NHCs [70,71]were also introduced into the ancillary ligand family (Scheme 1B).Anwander reported the synthesis of various alkylated carboxylate rare-earth complexes that catalyzedcis-1,4-polymerization of isoprene [72].These oxyanion ligands moved down a new path for the synthesis of dialkyl rare-earth complexes, whereas most of these complexes were not stable enough, presumably because of the easy ligand redistribution and complex aggregation reactions.Nevertheless, further studies about dialkyl rare-earth complexes bearing oxyanion ligands remain sluggish due to a lack of ligand diversity.Although some lanthanide carboxylates [76–79] and phosphates [80–85] were used as catalyst procursors in polymerization process, the well-defined alkyl complexes are yet undeveloped and the role of ligands is still obscure.Thus, we are quite curious about that if phosphates could be applied as anion ligands for dialkyl rare-earth complexes; a positive answer to this question could provide further insight about oxyanion ancillary ligands and also an attractive alternative catalyst.Herein, we reported the synthesis of well-defined phosphate yttrium dialkyl complexes and their catalytic application in stereo-controllable 1,4-polymerization of isoprene (Scheme 1C).
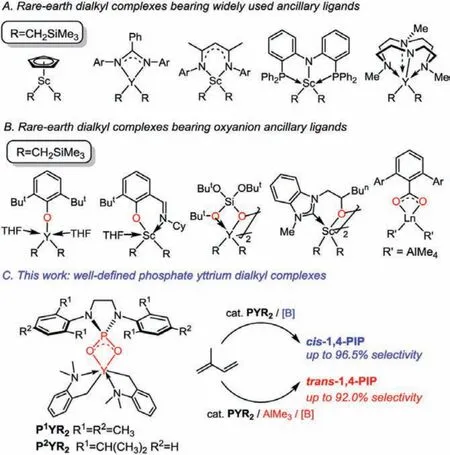
Scheme 1.Examples of rare-earth dialkyl complexes.
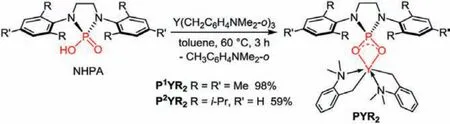
Scheme 2.Synthesis of phosphate yttrium dialkyl complexes.
The oxophilicity of rare-earth metals would definitely provide a strong interaction between the metal ion and the phosphate anion.This strong bonding, however, together with the small steric hindrance of the oxyanions, would probably result in the bridging interaction and ligand redistribution.To avoid the possible side reactions, we proposed to start with tris(aminobenzyl) yttrium complex, whose alkyl anion has a “built-in” chelating amino group.Thinking of the widely usedN-heterocyclic carbene ligands, we synthesized twoN-heterocyclic phosphorodiamidic acids(NHPAs) bearing similar skeleton structures and tunable steric parameters [86,87].The acid-base reaction between NHPAs and Y(CH2C6H4NMe2-o)3took place smoothly at 60 °C in toluene and afforded theN-heterocyclic phosphate yttrium bis(aminobenzyl)complexes PYR2after 3 h (Scheme 2).Both complexes were fully characterized by1H,13C and31P NMR spectroscopy, and their structures in solid state were determined by single-crystal X-ray diffraction (XRD).These characterizations reveal the similar structures of the two phosphate yttrium complexes, as shown in Fig.1.The phosphate anion is bonded to the yttrium center inκ2fashion through two oxygen atoms.The yttrium atom is well located on the OPO rigid frame to form a planar quadrilateral.In a simplified view, the phosphate ligand creates a well-defined steric pocket,along with theκ2coordination from the aminobenzyl ligands, conducing to enhance the stability of PYR2.Through comparison with the two phosphate yttrium complexes, it is showed that the YO bond lengths in P2YR2(2.306(2) Å) are longer than those in P1YR2(2.282(2) Å).We speculated that the larger steric hindrance from the isopropyl groups increases the distance between phosphate and yttrium atom.
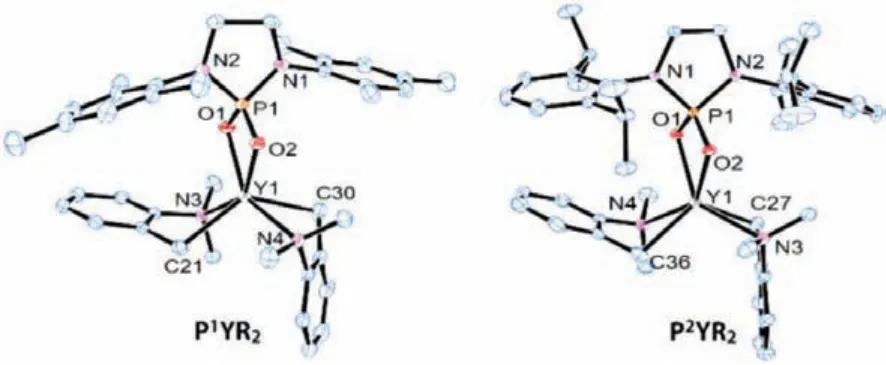
Fig.1.ORTEP structure of PYR2 complexes with thermal ellipsoids set at 30% probability.Hydrogen atoms and solvents have been omitted for clarity.Selected bond lengths [Å] and angles [°]:(P1YR2, left) Y1-O1 2.282(2),Y1-O2 2.277(2), Y1-C21 2.435(3), Y1-C30 2.435(3), Y1-N3 2.533(2), Y1-N4 2.522(2), O1-Y1-O2 63.63(8), P1-O1-Y1 96.19(1), P1-O2-Y1 96.12(1), O1-P1-O2 103.99(1).(P2YR2, right) Y1-O1 2.306(2), Y1-O2 2.300 (2), Y1-C27 2.439(3), Y1-C36 2.436(3), Y1-N3 2.525(2), Y1-N4 2.533(2), O1-Y1-O2 62.86(6), P1-O1-Y1 96.23(8),P1-O2-Y1 96.48(8), O1-P1-O2 104.30(1).
As part of our continuing investigations into isoprene polymerization, we then used the PYR2as catalysts for further exploration.The neutral P1YR2alone proved to be inactive for the isoprene polymerization (Table 1, entry 1).However, once activited with 1 equiv.of borate reagent [Ph3C][B(C6F5)4], P1YR2at room temperature demonstrated high catalytic activity:1000 equiv.of isoprene were converted quantitatively into PIP within 20 min (entry 2).Remarkably, the polymer product features a highcis-1,4 content (93.7%).GPC curve indicated that the PIP obtained is unimodal with aMnof 2.6 × 105and a narrow molecular weight distribution(PDI = 1.4).The catalytic polymerization under lower temperature took place smoothly and gave PIP with even highercis-1,4 content(96.5%) as well as higher molecular weight (Mn= 3.6 × 105) (entry 3).We next carried out the catalytic polymerization with various [Isoprene]/[P1YR2] ratios varying from 600 to 5000 (entries 4–9).All the reactions produced PIP with high efficiency (yields>98%) and excellent selectivity (cis-1,4 selectivity:92.6%-96.5%).Furthermore, the molecular weight of the resultant PIP increases linearly with the amount of the isoprene monomers (see Supporting information for details), while the molecular weight distribution still keeps narrow (1.4–1.6).The phosphate yttrium complex here displayed a very high catalytic activity and thus provided an approach for the synthesis of high-molecular-weightcis-1,4-PIP(Mnup to 1.06 × 106).Considering the important roles of alkylaluminum compounds in polymerization, we further carried out the catalytic polymerization reaction in the presence of trialkylaluminum reagents (entries 10 and 11).Al(iBu)3, a well-known chain transfer reagent, led to the decrease of the molecular-weight(Mn= 1.9 × 105), as expected.Unlike some known findings [46,48]that addition of AlMe3could increase thecis-1,4 content of PIP, the addition of 5 equiv.of AlMe3herein greatly increased thetrans-1,4 content (44.1%).
The catalytic system P2YR2/[Ph3C][B(C6F5)]4exhibited lowercis-1,4 selectivity in the polymerization of isoprene, but higher initiation efficiency (42.1%), compared to the catalytic system P1YR2/[Ph3C][B(C6F5)]4(25.9%), producing the PIP product with 83.3%cis-1,4, 5.3%trans-1,4 and 11.4% 3,4 contents (Table 2, entry 1).This result suggested that the steric effect of the twoorthosubstitutedN-aryl rings might be a contributing factor of thecis-1,4 selectivity.The bulkyo-isopropyl inN-aryl rings led to the decrease ofcis-1,4 selectivity [11,16].The catalytic system containing 5 equiv.of AlMe3surprisingly switched the stereoselectivityfromcis-1,4 totrans-1,4-polymerization, thus affording the polymers with contents of 89.4%trans-1,4 units (entry 6).Thetrans-1,4-polymerization took place much more slowly (90 min).The significantly increased molecular weight (Mn= 3.6 × 105) revealed an obvious decreased efficiency of the catalyst (18.7%), which hinted a dramatic structure change of the catalytic species.We speculated that the heteronuclear Y/Al complex formedin-situmight be the possible cataytic species in thetrans-1,4-polymerization [9,28,46].To find out more details, we carefully examined the polymerization with different [AlMe3]/[P2YR2] ratios.1 or 2 equiv.of AlMe3did not obviously change the regioselectivity (entries 2 and 3).Using 3 equiv.of AlMe3as additive resulted in a noticeable increase of thetrans-1,4 selectivity from 5.3% to 75.0% (entry 4).When 4 equiv.of AlMe3or more was added, the PIP was obtained with highertrans-1,4 content (85.1%-92%) (entries 5 and 7).It is suggested that 4 equiv.of AlMe3was necessary for the formation of the heterotrinuclear Y/Al complex [46].It is noteworthy that 10 equiv.of AlMe3as additive gave the PIP with lower molecular weight (Mn= 1.3 × 105) but similar narrow molecular weight distribution (PDI = 1.5), suggesting that the excess amount of AlMe3may act as a chain transfer reagent to interrupt the growing polymer chain and tune the molecular weight.For comparison, the PIP obtained with the system P2YR2/[Ph3C][B(C6F5)]4/AlEt3or Al(iBu)3failed to achieve hightrans-1,4 contents (entries 8 and 9).This observation showed that thetrans-1,4 selectivity might be influenced by the steric effect of the aluminum alkyls (AlMe3>AlEt3>Al(iBu)3) [21].Control experiments were conducted that the isoprene polymerization could not be initiated without either borate reagent or P2YR2, indicating that the cationic Y/Al species may account for thetrans-1,4-polymerization of isoprene (entries 10 and 11).To gain further insight into the possible heterotrinuclear Y/Al species, we carried out the reaction between phosphate yttrium dialkyl complexes and 4 equiv.of AlMe3.A crystal of a phosphate bis(tetramethylaluminate) yttrium complex [P1Y(AlMe4)2]2suit for X-ray diffraction analysis was surprisingly obtained (Fig.2).Compared to the crystal structure of P1YR2, this dimer complex[P1Y(AlMe4)2]2features a quite different coordination environment around the yttrium metal center.The huge change of the catalyst structure might result in the different coordination and insertion mode of isoprene, leading totrans-1,4-polymerization of isoprene (Table 1, entry 11).The synthesis and isolation of heteronuclear Y/Al complex [P2Y(AlMe4)2]2and catinoic species of[P1Y(AlMe4)2]2were tried but failed, possibly because of their thermal instability.
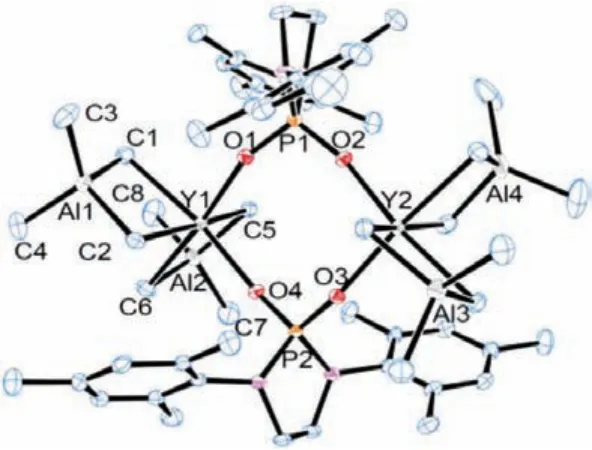
Fig.2.ORTEP structure of [P1Y(AlMe4)2]2 with thermal ellipsoids set at 30% probability.Hydrogen atoms and solvents have been omitted for clarity.Selected bond lengths [Å] and angles [°]:Y1-O1 2.188(2), Y1-O4 2.194(2), Y1-C1 2.621(4), Y1-C2 2.527(4), Al1-C1 2.062 (4), Al1-C2 2.074(4), O1-Y1-O4 95.883(8), O1-P1-O2 108.205(1), P1-O1-Y1 161.082(1), C1-Y1-C2 82.598(1), C1-Al1-C2 110.499(2), C3-Al1-C4 118.280(2).
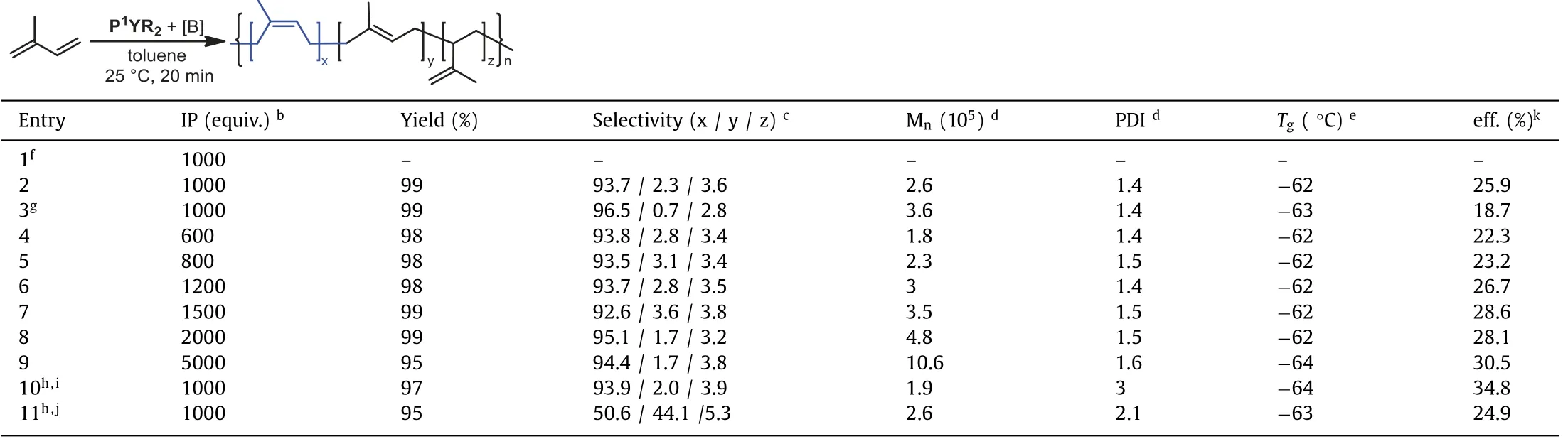
Table 1 P1YR2 catalyzed cis-1,4-polymerization of isoprene. a
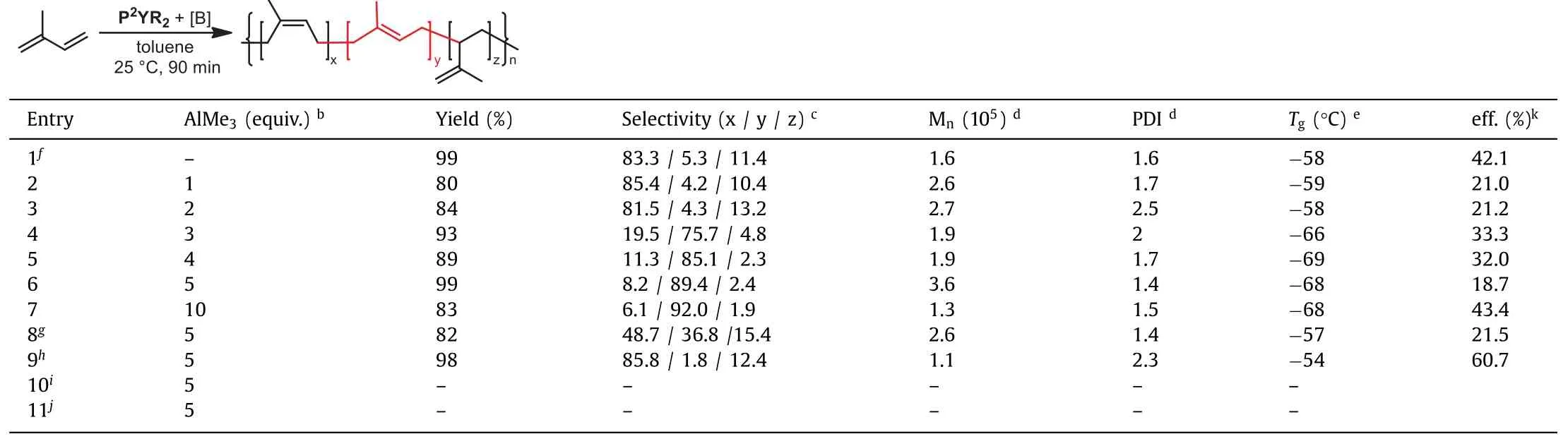
Table 2 P2YR2 catalyzed 1,4-polymerization of isoprene. a
In conclusion,N-heterocyclic phosphate ancillary ligand was designed and proved to be suitable for the synthesis and isolation of yttrium dialkyl complexes, which could be conveniently preparedviathe acid-base reactions between tris(aminobenzyl) yttrium complex andN-heterocyclic phosphoric acid.Activated by[Ph3C][B(C6F5)]4, the yttrium complexes PYR2exhibit high catalytic activities forcis-1,4-selective (up to 96.5%) polymerization of isoprene.The stereoselectivity was dramatically switched fromcis-1,4 totrans-1,4-polymerization (up to 92.0%) when using AlMe3as an additive.Considering various easily available phosphoric acids including the chiral ones, we could expect a nice synthetic value and diversified catalytic applications of phosphate ligated metal complexes in near future.Along this line, the synthesis ofNheterocyclic phosphate complexes with other metals and also with chiral phosphoric acids are currently ongoing in our laboratory.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We gratefully acknowledge the support from the Fudan University.We thank Prof.Li Pan (Tianjin Univ.), Prof.Jian-Hua Cheng(CIAC, China) for their helpful suggestions.This project was supported by National Natural Science Foundation of China (Nos.21890722 and 21950410519), National Natural Science Foundation of Tianjin (No.19JCYBJC20100).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.064.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction