
Palladium-catalyzed relay C–H functionalization to construct novel hybrid-arylcyclophosphorus ligand precursors
2022-06-20 07:59JuanWangPengBoBaiShangDongYang
Chinese Chemical Letters 2022年5期
Juan Wang, Peng-Bo Bai, Shang-Dong Yang
a State Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, China
bState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000,China
Keywords:Palladium catalyst Cyclization Acetylation Hydroxylation Dibenzophosphorus oxide
ABSTRACT A new relay C–H functionalization of di([1,1′-biphenyl]-2-yl)phosphine oxide to obtain esterified and hydroxylated products with different hypervalent iodines as oxidants under palladium catalysis is disclosed.This reaction provides a more effective and concise strategy for the synthesis of novel structural hybridarylcyclophosphorus ligand precursors with a wide range of substrates and good functional group tolerance.
Biaryl phosphorus oxides are a class of important precursors of phosphine-based ligands [1–9].They are often modified to obtain various hybrid phosphorus compoundsviadifferent strategies[10–14].Recently, cyclic aryl phosphorus oxides have also received much attention for their special properties in fluorescence materials [15–21].Therefore, searching for a concise and efficient strategy to synthesize various new structural biaryl phosphorus oxides and derivatives is required, which is currently considered a hot topic[22–36].It is evident from recent literature that C–H activation has emerged as an active field of research [37–50].In this regard,various aromatic C–H functionalizations involved in diphenylphosphorus oxides have become a most fashionable synthetic strategy [51–56].In 2011, Taki and coworkers reported an intramolecular dehydrogenative cross-coupling of diphenylphosphine oxides through the palladium-catalyzed C–H activation [57].In addition,transition-metal catalyzed (2-bromophenyl)diphenylphosphine oxide C–H cyclization also offers a practical pathway [58–61].Meanwhile, the radical-initiated intermolecular [3 + 2] cycloaddition of diphenylphosphine oxides with alkynes, by C–H activation, to structure biaryl phosphorus oxides has been reported by various research groups [62–72].
Over the past several years, significant progress has been made by various research groups looking into R2(O)P-directed C(sp2)-H functionalization [73–84].Subsequently, we selected[1,1′-biphenyl]-2-yldiphenylphosphine oxides as substrates to realize various C–H functionalizations [85–89], including esterification and hydroxylation (Scheme 1a) [90–91].We successfully achieved a new relay C–H functionalization of di([1,1′-biphenyl]-2-yl)phosphine oxide to obtain esterified and hydroxylated products with different hypervalent iodines as oxidant under palladium catalysis (Scheme 1b).This reaction provides a more effective and concise method for the construction of novel hybridarylcyclophosphorus ligand precursors.
Initially, we selected di([1,1′-biphenyl]-2-yl)phosphine oxide(1a) as a model substrate under the catalysis of Pd(OAc)2(10 mol%), and with PhI(OAc)2as both the source of acetate and oxidant, in trifluoroethanol solvent, to carry out the reaction at 100 °C (Table 1).To our delight, we obtained the desired product with a yield of 51% when 2.0 equiv.PhI(OAc)2was used (entry 1).Encouraged by this result, we proceeded to further optimize the reaction conditions.First, we screened different solvents and found that trifluoroethanol remained the best choice (entries 2–5).TLC monitoring showed formation of the intermediate 5-([1,1′-biphenyl]-2-yl)benzo[b]phosphindole 5-oxide (m, Scheme 1).Thereafter, it was determined that for this transformation a higher temperature was favorable; the yield of product could be increased to 62% at 120 °C (entries 6 and 7).Following on from this, different oxidants, and their content, were carefully screened.The amount of PhI(OAc)2is crucial to the successful C–H esterification.When 4.0 equiv.PhI(OAc)2was added to the reaction system, the yield of product improved to 92% (entries 8 and 9).Further increasing the PhI(OAc)2to 5.0 equiv.did not afford any higher yield (entry 10).Several other oxidants were also investigated (entries 11 and 12).When using oxane in the reaction, only 35% of the product was obtained.Finally, different palladium catalysts, and their content,were further screened.It emerged that Pd(OAc)2was most favorable here (entries 13–20).
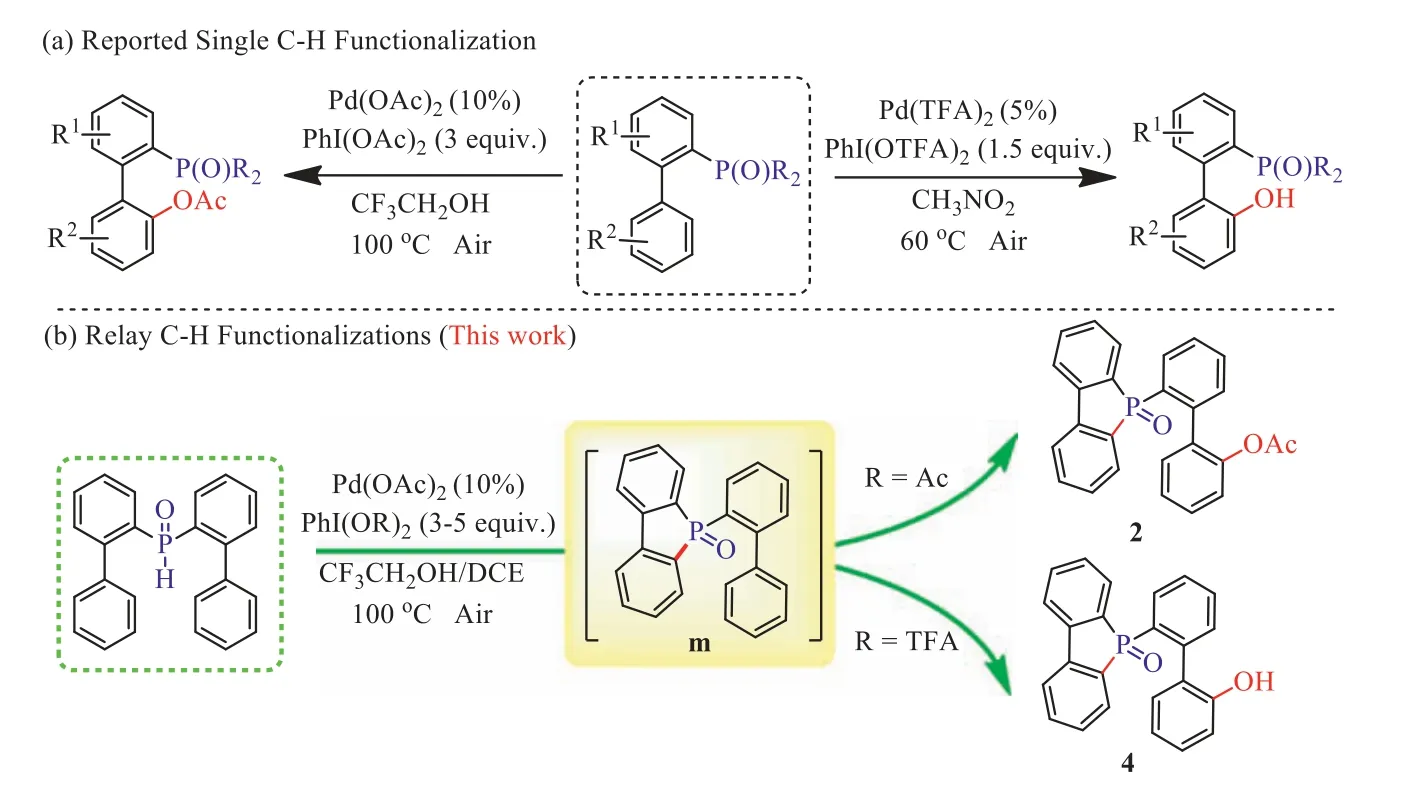
Scheme 1.P=O directed C–H esterification and hydroxylation.
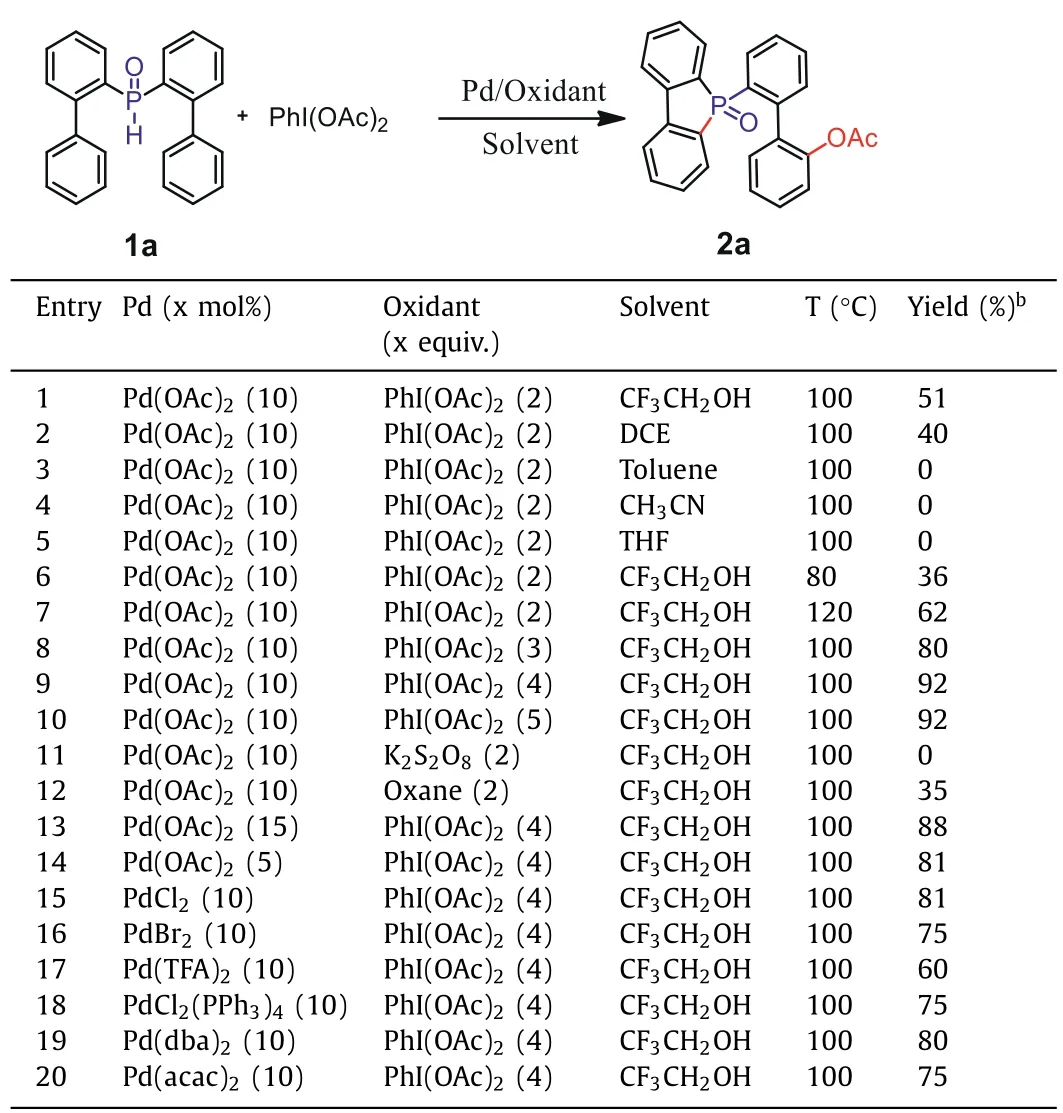
Table 1 Optimization of reaction conditions.a
We then investigated the range of substrates, under the optimal reaction conditions, for different substituents (Scheme 2).First, we considered the aromatic ring on the biphenyl group that is not connected to P–H (2b–2e).When one or more electrondonating methyl groups are located at different positions of the aromatic ring, the reaction can proceed smoothly, and medium to high yields can be obtained.It is evident that steric hindrance does not have much effect on it.When a substrate with a strong electron donor (–OMe) is added to the reaction, we achieve a higher conversion rate and obtain a yield of 80% (2f).However, when the substituent is a phenyl group, the yield decreases (2g).Disappointingly, when the electron-withdrawing group is in theparaposition,the yield is significantly reduced (2h, 2i).It is evident that the electronic effect has a great influence; the electron-withdrawing group is not conducive to the conversion in this reaction.When the substituent is changed to an aromatic ring connected to P–H,the result of the electronic effect is also obvious.It further proves that the conversion effect of the electron-donating group is obviously beneficial to the electron-withdrawing group (2j, 2k).Simultaneously, when all the aromatic rings have electron-donating substituents, the yield is also good (2l, 2m).We successfully recrystallized product 2a as colorless crystals and were able to confirm the molecular structure by X-ray diffraction analysis.
Inspired by the success of the above esterification and results recorded in our previous work [12], we proceeded to further realize the hydroxylation using the same strategy.Thus, we investigated the use of different hypervalent iodines, solvents, and reaction temperatures, with Pd(OAc)2as catalyst.As expected, the hydroxylated product was successfully obtained in 51% yield when using [bis(trifluoroacetoxy)-iodobenzene] (PhI(OTFA)2) as oxidant in 1,2-dichloroethane (DCE) at 60 °C.We then conducted a specific screening of the reaction conditions and obtained the optimal conditions (for details see Supporting information).Subsequently,we conducted research on different substrates and examined the scope and limitations of this method (Scheme 3).First, the substituents on the aromatic ring not connected to P–H and the steric hindrance effect were investigated.When it contains one or two electron-donating groups, the substrate has better tolerance and a higher yield can be obtained (4a–4c).Unfortunately, when the aromatic ring has a strong electron-withdrawing group, the target product cannot be obtained (4d).When the position of the substituent is changed to the aromatic ring connected with P–H,both the electron-donating and electron-withdrawing substituents can react; however, the yields of the electron-withdrawing products are obviously not as good as those of electron-donating products.It is evident that the electronic effect has a greater impact on the range of the substrate (4e–4g).
During the course of the substrate investigations, we found that the electron-deficient groups such as F, CF3andnBuCO lay in any aromatic ring, only the cyclization occurred and the corresponding product of 6a-6d was obtained in good yields (Scheme 4).These results indicated that the electronic effect has a greater impact on the C-H esterification or hydroxylation.
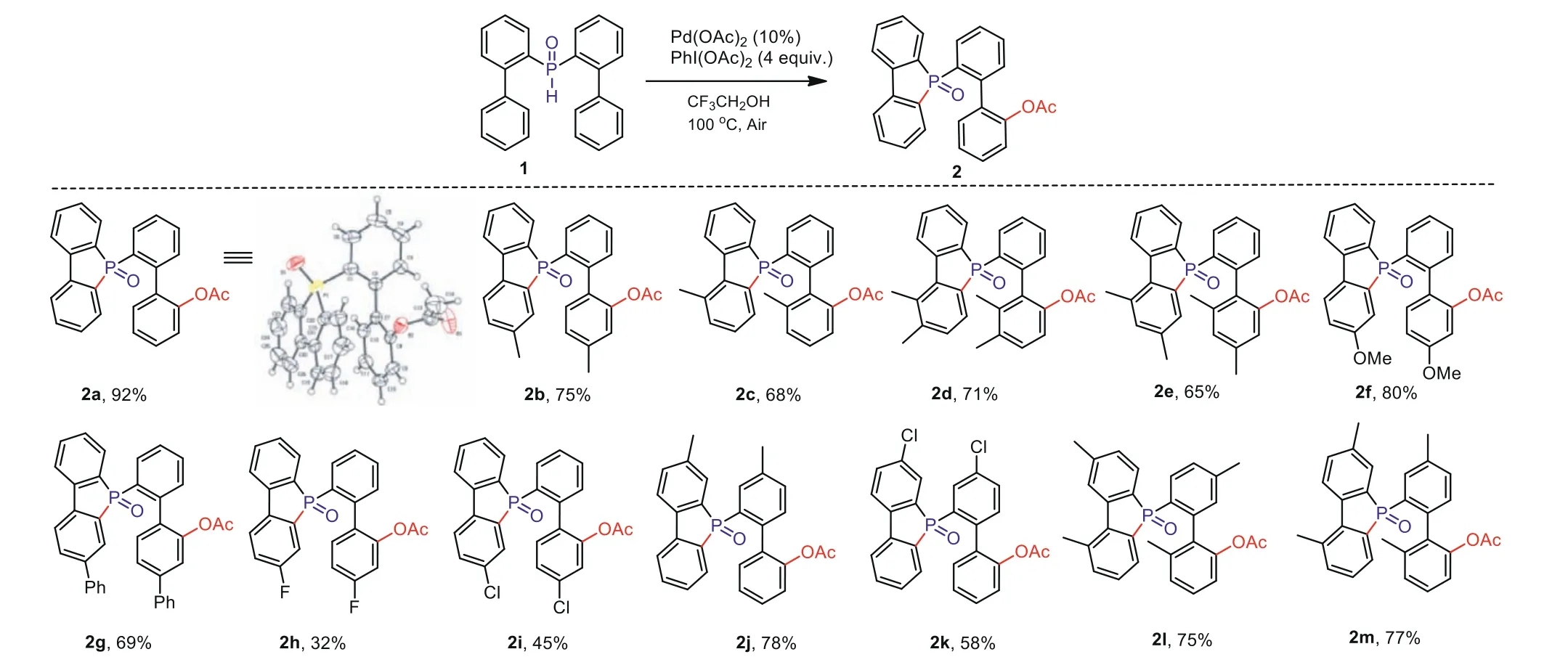
Scheme 2.Substrate scope of the acetoxylation.Reaction conditions:1 (0.1 mmol), PhI(OAc)2 (4.0 equiv.), Pd(OAc)2 (10 mol%), CF3CH2OH (1.0 mL), air atmosphere, 100 °C.Isolated yields of products.
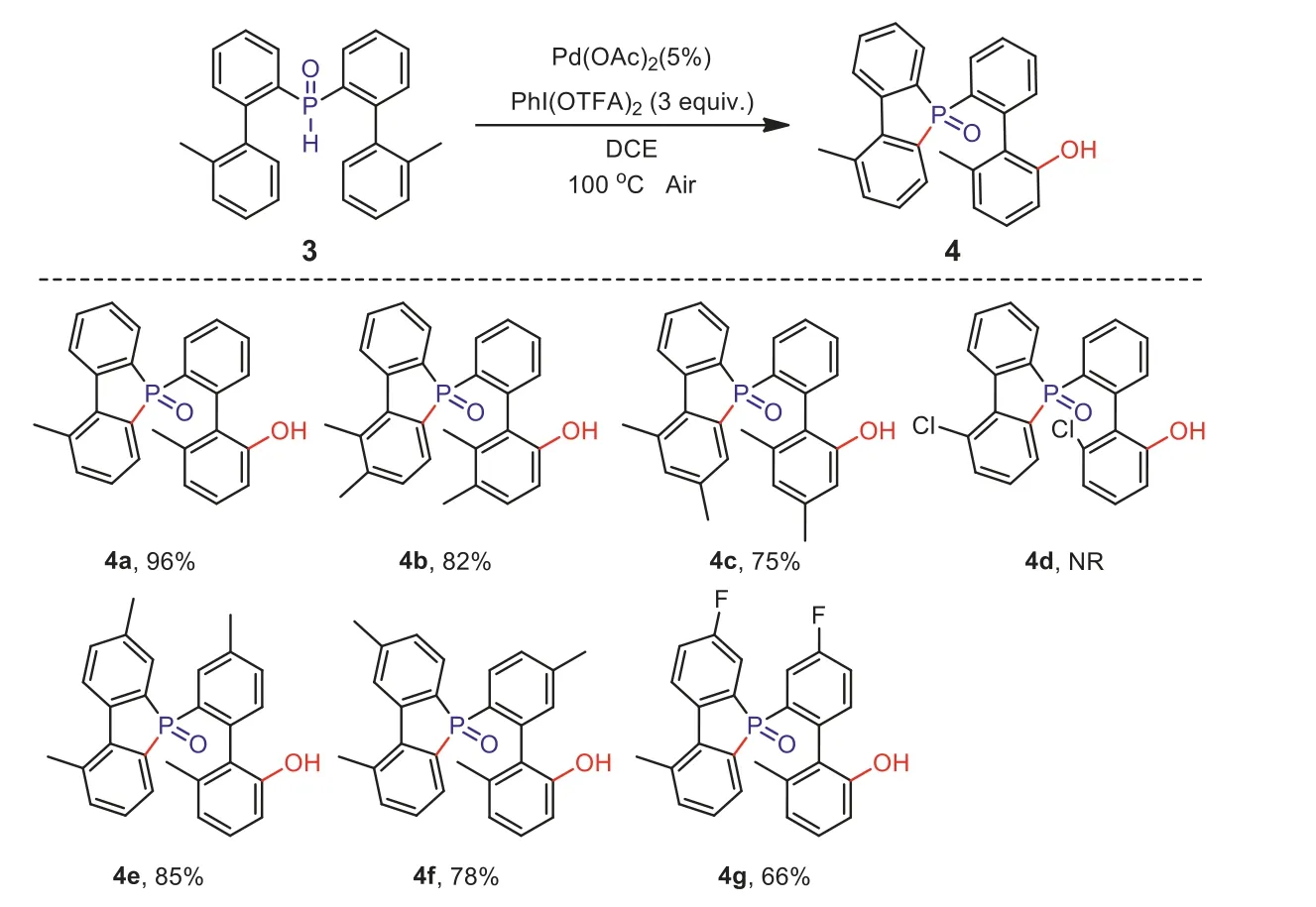
Scheme 3.Substrate scope of the hydroxylation.Reaction conditions:3 (0.1 mmol), PhI(OTFA)2 (3.0 equiv.), Pd(OAc)2 (5 mol%), DCE (1.0 mL), air atmosphere, 100 °C.Isolated yields of products.
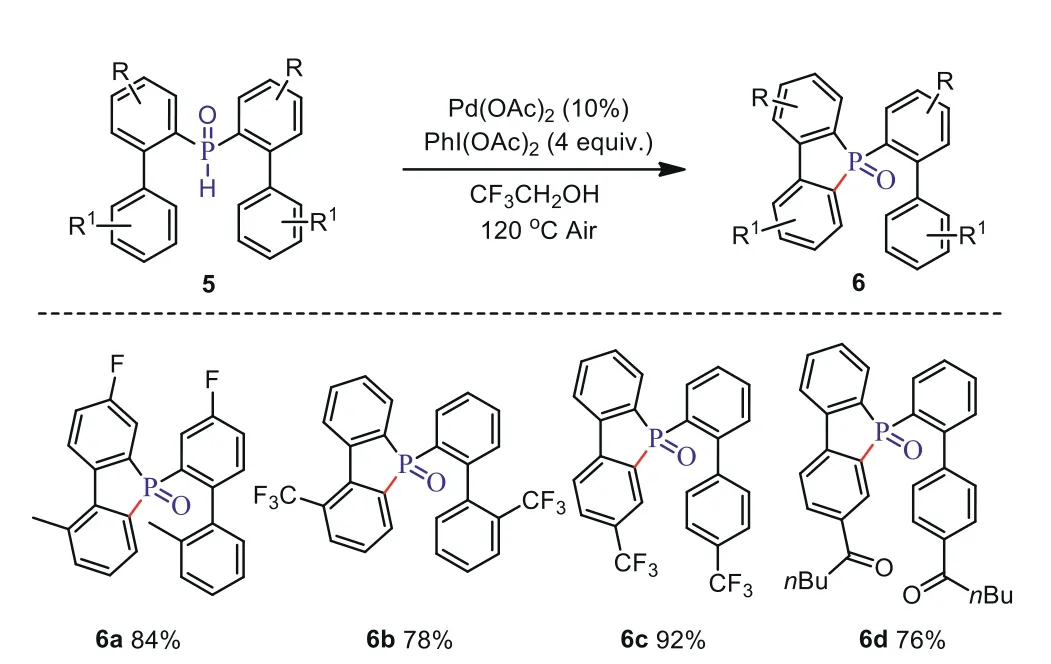
Scheme 4.Scope of the electron-deficient substrate.Reaction conditions:1(0.1 mmol), PhI(OAc)2 (4.0 equiv.), Pd(OAc)2 (10 mol%), CF3CH2OH (1.0 mL), air atmosphere, 120 °C.Isolated yields of products.
In the cross-coupling reaction involving transition metals,monophosphorus ligands are important participants.Therefore, we adopted a classic reduction system for its standard products.Fortunately, the reduced product was obtained in a higher yield.Simultaneously, it had been done a grading reaction, the results of which were also very impressive (Scheme 5).
To demonstrate the utility of our ligands, we used the reduced product of 5a as a ligand to promote the Pd-catalyzed different cross-coupling reactions (Scheme 6).It exhibited the excellent activity in the Suzuki-Miyaura, Heck and Sonogashira coupling reactions and the corresponding coupling product was afforded in the best yield.
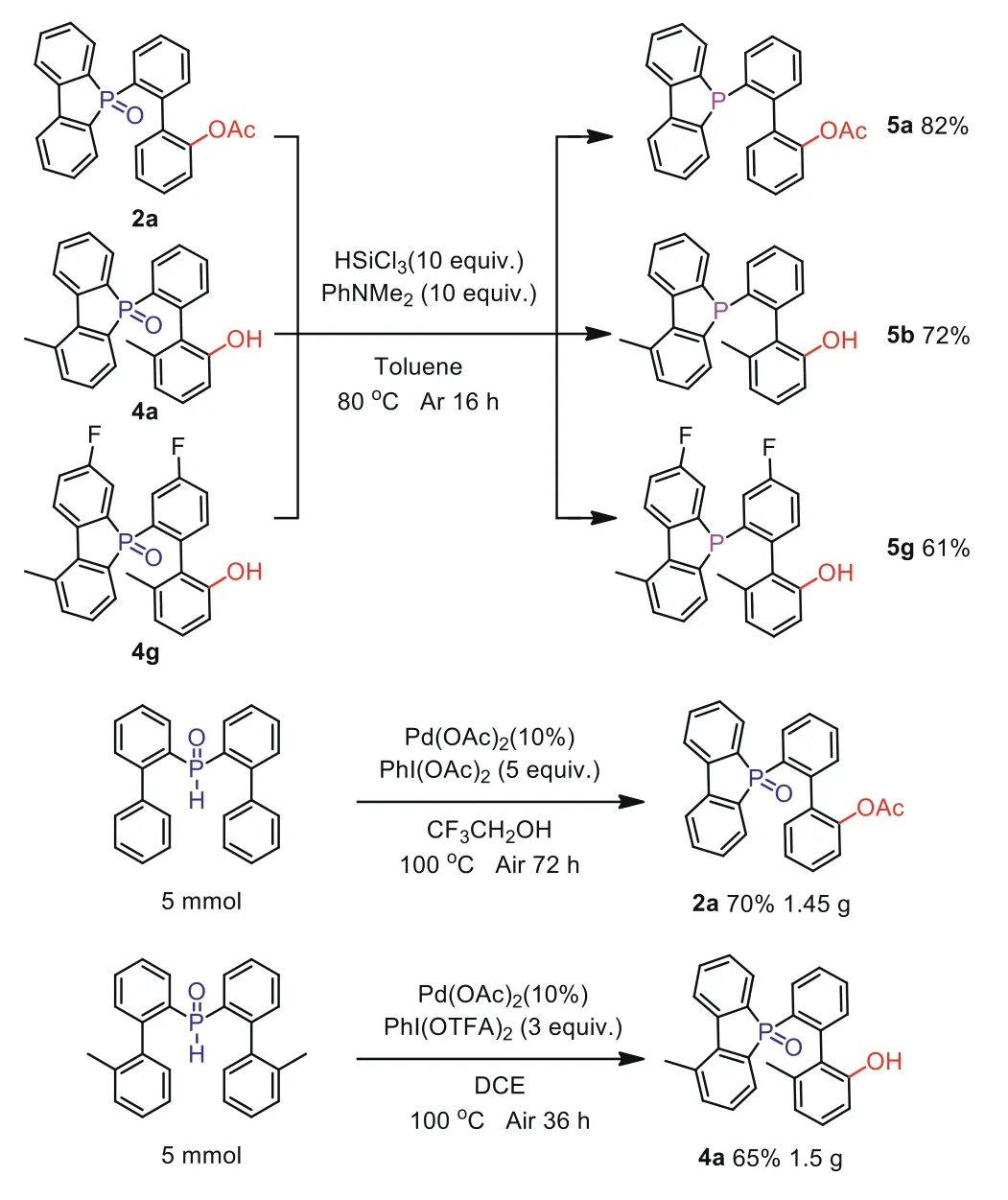
Scheme 5.Reduction of phosphorous oxides to monophosphorus ligands and gram-scale experiment.
To then further address the reaction mechanism, we conducted control experiments (Scheme 7).First, when Pd(OAc)2is not added under standard conditions, 92% of the intermediate cyclic phosphorus product m can be obtained.Based on the above conditions,when radical scavengers (TEMPO) are added, the reaction will not occur.Under standard conditions, the intermediate product m can be obtained without adding an oxidant or adding TEMPO at the same time.It is then evident that the activation of the C–H bond in the first step is a coordinated process of Pd(OAc)2and the oxidant.
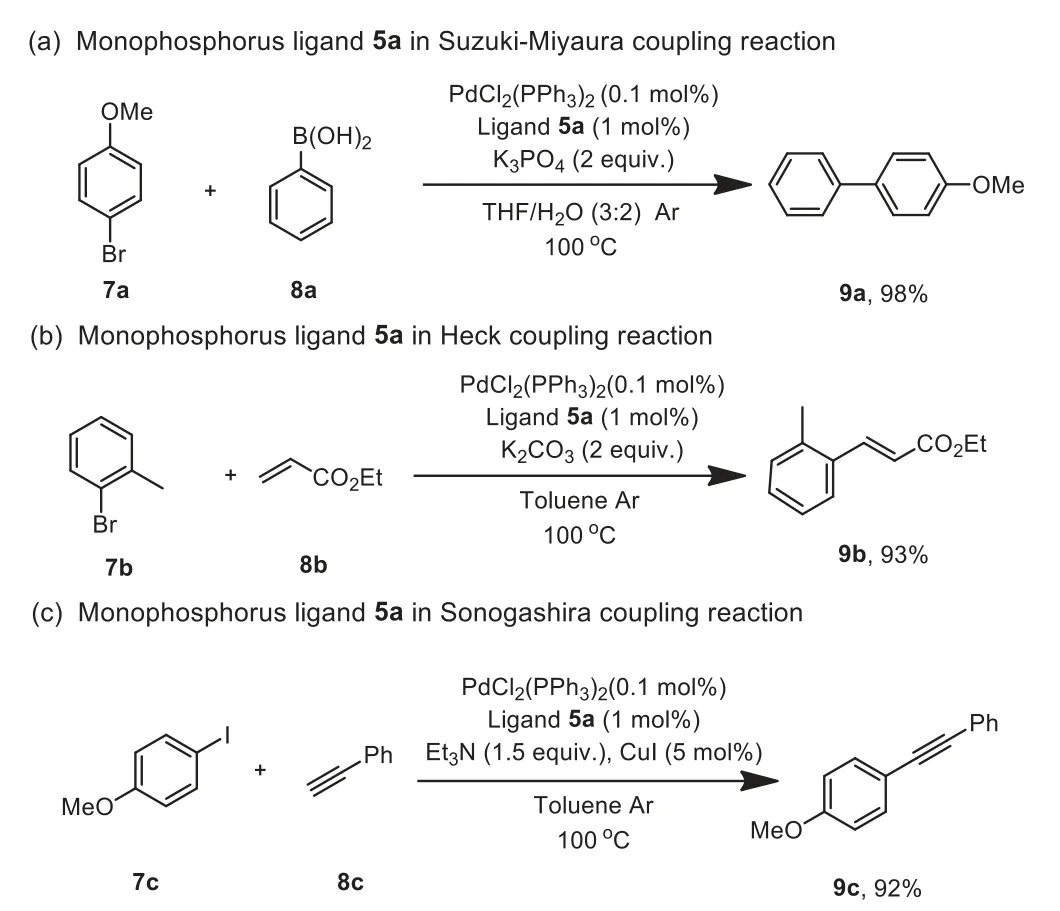
Scheme 6.Synthetic application.Reaction conditions:7 (0.5 mmol), 8 (0.65 mmol),PdCl2(PPh3)2 (0.1 mol%), ligand 5a (1 mol%), and base in solvent (2 mL) at 100 °C under argon, 10 h.
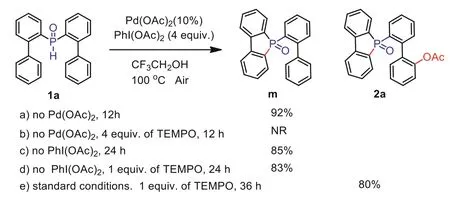
Scheme 7.Mechanistic aspect:Control experiments.
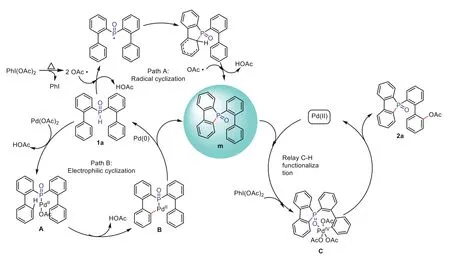
Scheme 8.Proposed reaction mechanism.
According to results of the above experiments and previous literature reports [57], the possible pathway of this relay C–H functionalization is proposed, as shown in Scheme 8.Initially, the first step of C–H bond cyclization involves two paths, namely the Pudovik reaction process of radicals and the catalytic cycle between Pd(II) and Pd(IV) species.The hypervalent iodine oxidant generates radicals under heating conditions, initiating the activation of the C–H bond of the P radicals, and thereby generating the dibenzophosphorus oxide intermediate m (Path A).Meanwhile, the substrate 1a undergoes oxidative addition, C–H bond activation,and reductive elimination under palladium catalysis to afford the dibenzophosphorus oxide intermediate m (Path B).Subsequently,it is guided by P=O through the 7-membered ring palladium intermediate to then perform the second step in the C–H bond activation, finally affording the desired esterified and hydroxylated products.
In conclusion, we have developed a novel method for the esterification and hydroxylation of dibenzophosphorus oxide by relay C–H functionalization.The procedure shows good functional group tolerance, and the corresponding products are obtained in high yield and with high selectivity.We envisage that this reaction will offer an effective method for the synthesis of different functionalized diphenyl phosphorus oxides.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful to the National Natural Science of China (No.21532001) and the International Joint Research centre for Green Catalysis and Synthesis (No.2016B01017) and Lanzhou University for their financial support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.10.019.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction