
MnBr2 catalyzed regiospecific oxidative Mizoroki-Heck type reaction
2022-06-20 07:59XiangChenZhihongZhuShanshanLiuYiHungChenXiaoShen
Chinese Chemical Letters 2022年5期
Xiang Chen, Zhihong Zhu, Shanshan Liu, Yi-Hung Chen, Xiao Shen
Institute for Advanced Studies, Engineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, Wuhan University, Wuhan 430072, China
Keywords:Fluoroalkylation Mizoroki-Heck Mn catalysis Radical 1,2-silyl transfer Homoallylic alcohols
ABSTRACT Herein, we report an unprecedented regiospecific oxidative Mizoroki-Heck type reaction for the synthesis of α-difluoromethyl homoallylic alcohols.The reaction shows broad substrate scopes and high functional group tolerance.Late-stage functionalization of complex biologically active molecules demonstrates the synthetic potential of this transformation.Mechanistic study supports the involvement of MnBr2 catalyzed radical 1,2-silyl transfer.
Olefins are not only one class of important synthetic building blocks in chemistry but also common structural motifs of pharmaceuticals, natural products, organic materials [1–4].Therefore,preparation of functionalized alkenes is one synthetic transformation pursued by chemists for a long time [5–9].Mizoroki-Heck reaction is a well-known C–C bond formation reaction involving carbometallation andβ-hydride elimination pathway [10–18].However, the key challenge remained is the regioselectivity control to generate terminal or internal alkenes, because both Haand Hbare prone to be eliminated (Scheme 1a) [19–27].Moreover, internal olefins are thermodynamically more stable, therefore the kinetically controlled synthesis of terminal olefins are extremely difficult because of possible post-isomerization [22,28-31].Recent progress made by Nishikata group through Cu-catalyzed Mizoroki-Heck type reaction afforded mostly 91/9 (terminal alkene/ internal alkene)selectivity, but theZ/Eselectivity in the synthesis tri-substituted olefin is low (64:36,E/Z) [32–34].
Fluorinated molecules often show improved lipophilicity, solubility, and metabolic stability than the parent compounds [35].Among them, difluoromethylated compounds are of particular interests, because CF2H could mimic OH and SH and it is also known to be a hydrogen donor through hydrogen bonding [36–42].For example, difluoromethylated alcohols such as anti-tumor agent A[43] and cathepsin K inhibitor B [44] have gained increasing attention in pharmaceutical and life-related sciences (Scheme 1b).In 2020, our group [45] reported a synthesis of fluoroalkyl substituted homoallylic alcohol C enabled by radical 1,2-silyl transfer [46–52].Although the yield is synthetically useful, the allylic sulfone substrate is needed to be synthesized fromα-methyl styrene [45].Herein, we disclose an efficient synthesis of difluoromethylated homoallylic alcohols directly fromα-alkyl styrenes through MnBr2catalyzed regiospecific oxidative Mizoroki-Heck type reaction with our difluoromethylated organosilicon reagent (Scheme 1c).The new methodology is more step and atomic economical than the previous method [45].The present allylic C–H alkylation reaction is operationally simple and shows broad substrate scopes along with high functional group tolerance.The late-stage functionalization of bioactive complex molecules and a gram scale reaction demonstrate the synthetic potential of our methodology.
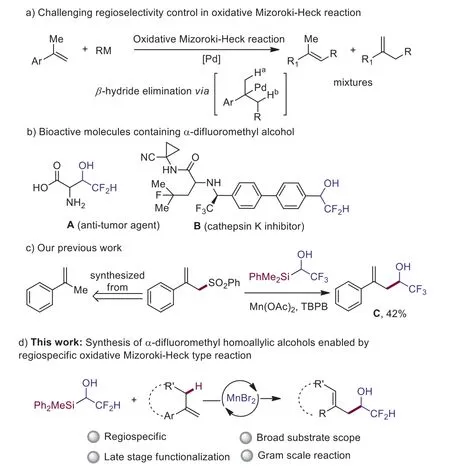
Scheme 1.Background and our strategy in the synthesis of difluoromethyl homoallylic alcohols.
We investigated the reaction with 1a andα-methyl styrene 2a as model substrates under catalytic system of MnBr2/TBPB in DCM,and the target product 3a was obtained in 48% yield after desilylation with TBAF (Table 1, entry 1).Encouraged by the result of selective formation of terminal alkene 3a, we further investigated the influence of ligands on the efficiency of the reaction (Table 1,entries 2–5).The use of tridentate L1 or bidentate L2 as the ligand resulted in no formation of product 3a (Table 1, entries 2 and 3).However, 2,2′-bipyridine type ligands are better choices.With L3 as a ligand, 65% yield was observed (59% isolated yield, Table 1,entry 4).When L4 was employed as the ligand, the yield of product 3a increased to 68% (67% isolated yield, Table 1, entry 5).Altering silyl group from SiMe2Ph to SiMePh2, the efficiency of reaction was further improved and 3a was formed in 73% yield (isolated in 76% yield), but the yield could decrease to 53% without L4 as the ligand (Table 1, entry 6).Other silyl group such as SiPh3, SiEt3,SiMe3substituted substrates 1c~1e afforded lower yield (Table 1,entries 7–9), but SiiPr3and SiPh2tBu substituted reagents 1f and 1g could not afford product 3a owing to the steric hindrance of the silyl groups (Table 1, entries 10 and 11).Control experiments showed both catalyst and oxidant are important for the success of this reaction, since no conversion of the organosilicon reagent was observed without either MnBr2/L4 or TBPB (Table 1, entries 12 and 13).
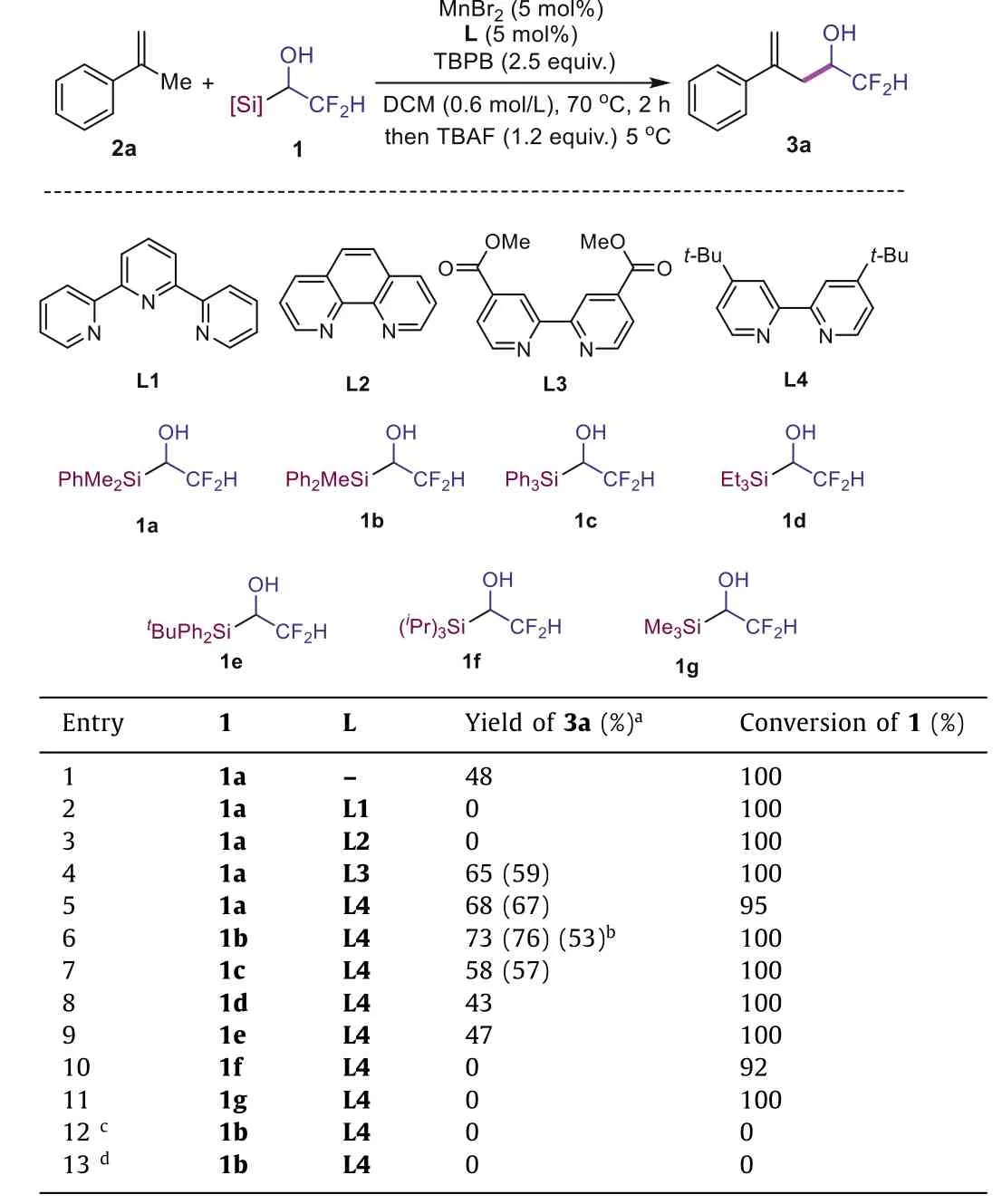
Table 1 Investigation of reaction conditions.
With the optimized condition in hand, we next explored the scope of reaction.We found a variety ofα-alkylstyrenes bearing different functional groups could be used as substrates, affording theα-difluoromethyl substituted homoallylic alcohols 3b-3ad in 46%-77% yields (Scheme 2).This reaction can be scaled up,and compound 3a was obtained in yield of 66% (1.31 g isolated).Many functional groups could be tolerated.Csp2-X (X= F,Cl, Br,I) bond was maintained after the reaction, affording product 3e-3h in 58%-65% yields.Moreover, OMe, OAc, OCF3and SMe could be tolerated, giving corresponding products 3c, 3i-3k in 50%-76%yields.An amide containing product 3l was also isolated in 71%yield.Mutiple substituted styrenes were used as substrates to generate products 3n and 3o in 51% and 71% yield.Biaryl and naphthlenyl substituted olefins are also suitable substrates, resulting in products 3q-3u in 54%-68% yields.Heterocyclic groups, such as benzofuryl, benzothienyl groups were also tolerated by the current oxidative reaction conditions (3v, 46%, 3w, 46%).A variety of ethers could also generate desired products (3x 64%, 3y 54%, 3z 61%, 3aa 64%).It was worth noting that phenol product 3ab could be obtained in 58% yield with 4-OTBS substituted styrene as starting material.To our delight, tri-substituted alkene containing hydroxyl difluoroalkyl group such as 3ac was successfully synthesized in 66% yield.Notebly, compounds 3ad was isolated in 72% yield as single regioisomer and stereoisomer, indicating the excellent regio and stereoselectivity of our reaction.
To further evaluate the efficiency of the regiospecific oxidative Mizoroki-Heck type reaction, we next applied this protocol in late-stage synthesis of relatively more complex molecules.When testosterone derived terminal olefin 4a was emplyed as the substrate in the formal C–H alkylation reaction, corresponding product 5a was obtained in 68% yield (Scheme 3).It was worth noting that N–H bond in substrate 4b could also be tolerated in the reaction,affording product 5b in 42% yield, which is an obvious advancement over our previous work, because N–H bonds were detrimental to the reaction of allylic sulfones [45].Moreover, estrone and estradiol derivedα-methyl styrenes were also effective in present condition, affording desired alcohols 5c and 5d in 60%-70% yields.In addition, the reaction of proline derived ester 4e was also effi-cient, affording product 5e in the yield of 48% (Scheme 3).
Several control experiments were conducted to probe possible mechanism.Firstly, we found that the MnBr2catalyzed oxidative Mizoroki-Heck type reaction was inhibited by the addition of radical trapping agent 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO),and compound 6 was detected by HRMS (Scheme 4a).Therefore,radical process is likely involved in current reaction.The detection of compound 7 (Scheme 4a) and homo-coupling product 8 in the reaction of 2a under standard condition (Scheme 4b) led us to consider the possibility of the radical-radical coupling between allyl radical and ketyl radical (for details, see Supporting information).However, the deuterium-incorporation experiment did not support this possibility because compound 3q-d2was isolated in 51% yield, when compound 2q-d3was used as the substrate (Scheme 4c).There was no deuterium-incorporation in the allylic position of compound 3q-d2(Scheme 4c).Moreover, competitive reaction between 2q and 2q-d3showed that KIE was 1.04, which implied the deprotonation-elimination process was not the rate-determining step (Scheme 4d) [52].We have shown that Mn(II) catalyst is important for the success of the reaction (Table 1,entry 12).Further investigation indicates that bromide ion is also crucial (Scheme 4e).We found that when Mn(OAc)2was used as the catalyst, no product 3a was generated although the organosilicon reagent was consumed in about 20%.However, the addition of LiBr (10 mol%) led to the formation of compound 3a in 35%yield, while Mn(OAc)2(5 mol%) was used as the catalyst.Similar trend was found for the catalysis with MnSO4·H2O (without LiBr,0% yield of 3a, 10% conversion of 1b; with 10 mol% LiBr, 30% yield of 3a, 75% conversion of 1b).While there is 21% yield of 3a formed when MnCl2·4H2O was employed as the catalyst, the addition of LiBr also significantly increased the efficiency of the reaction (69%yield of 3a, 100% conversion of 1b).Allylic bromide was ruled out as the intermediate in the present protocol because no product 3a was detected when 9 was added to the reaction (Scheme 4f).
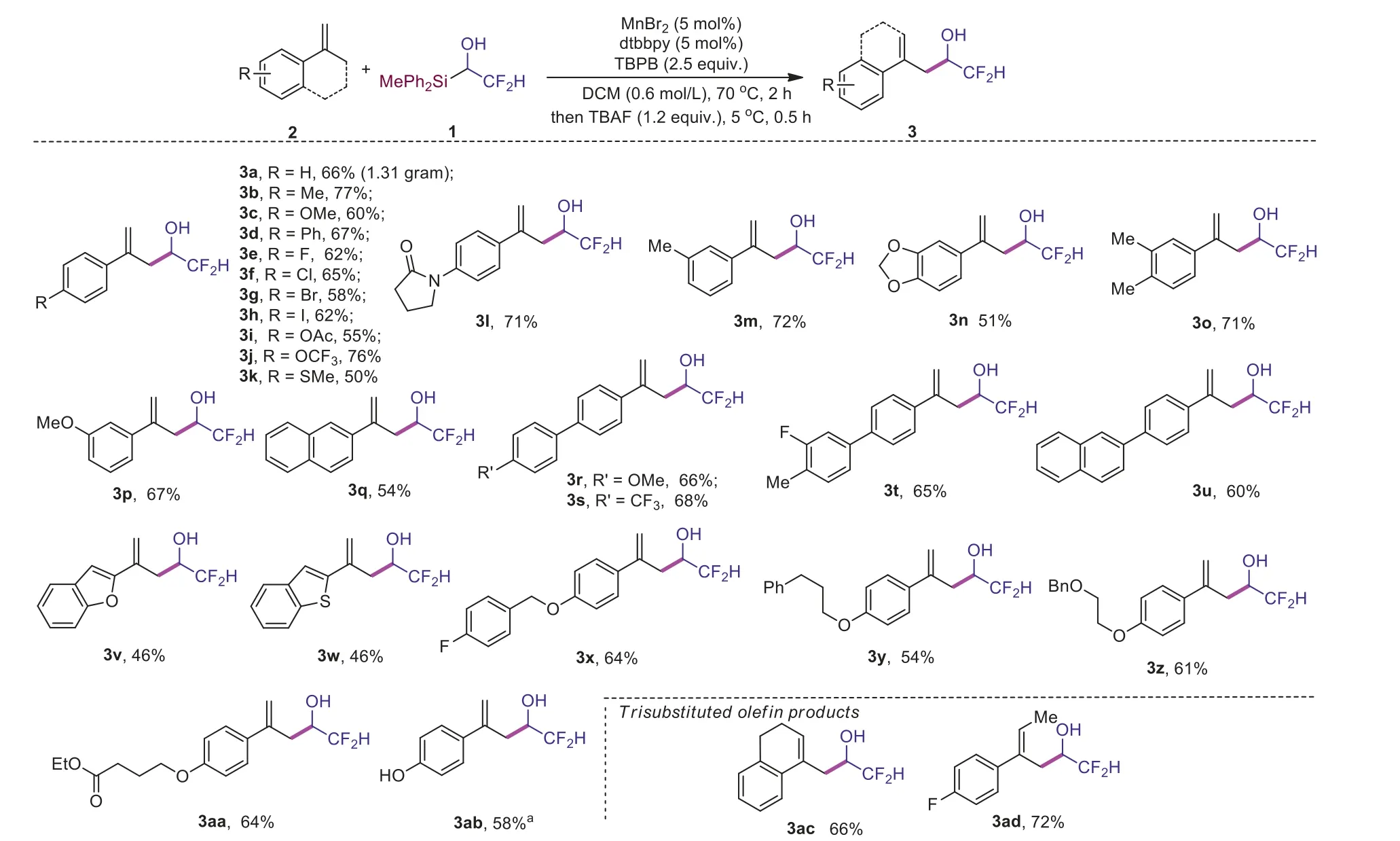
Scheme 2.Investigation of substrate scopes.Reactions were conducted under standard condition and isolated yield was provided for each product.a 4-OTBS substituted reagent 2aa was used.
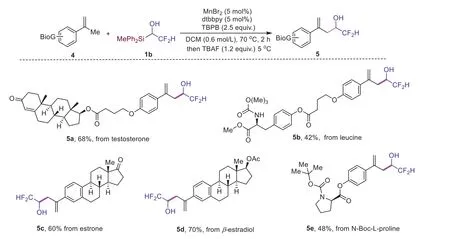
Scheme 3.Late-stage functionalization of alkenes derived from bioactive complex molecules.
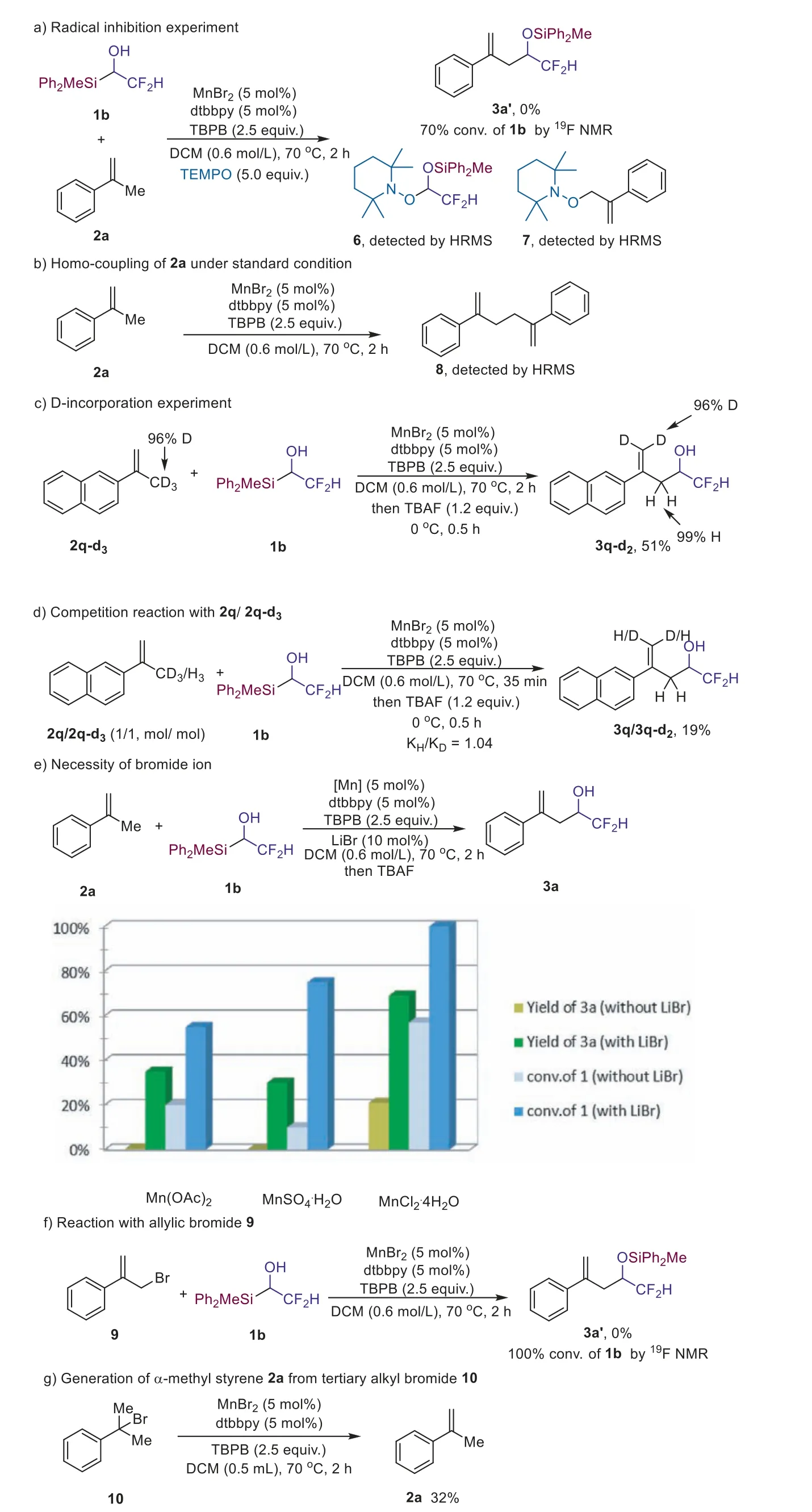
Scheme 4.Control experiments and mechanism study.
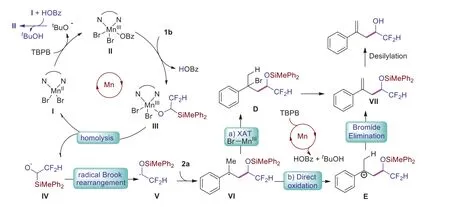
Scheme 5.Plausible mechanism for product formation.
Since bromide is important for the success of the reaction,we propose that intermediate D might be involvement in the reaction (Scheme 5).We indeed found thatα-methyl styrene 2a could be easily generated from tertiary alkyl bromide under the standard reaction (Scheme 4g).The KIE experiment indicate that the deprotonation-elimination step might not the rate-determining step which could explain why we could not isolate intermediate D.Based on the above experimental results and previous report[32-34,45,53], a plausible mechanism was proposed (Scheme 5).Mn(II) could be oxidized to Mn(III) by TBPB, and intermediate III might be formed by an anion exchange process, which could undergo homolysis to generate the alkoxyl radical IV.Thein-situgenerated flourinated ketyl radical Vviaradical Brook rearragement was added to styrene to form the new benzyl radical VI, and intermediate D could be generated through halogen atom abstraction(XAT) from liganted Mn(III) bromide.The selective deprotonative bromide elimination process from the less sterically hindered side would afford the olefin product (Scheme 5).However, we could not rule out the possibility of the formation of productviaintermediate E which would be generated through direct oxidation by hypervalent Mn species.
In conclusion, we have developed a MnBr2catalyzed regio- and stereo-selective oxidative Mizoroki-Heck type reaction.Various difluoromethylated homoallylic alcohols have been produced in synthetically useful yield.The broad applicability and general utility of this formal C–H alkylation reaction is demonstrated by the wide substrate scopes and highlighted by the late-stage introduction of the difluoroalkyl group to natural product derived olefins.The observed selectivity was proposed to be resulted from the bulky alkylation reagent.We believe that the finding of the selectivity control in this work will attract chemists’interest in the application of oxidative Mizoroki-Heck type reaction in the synthesis of functionalized terminal alkenes.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We are grateful to the National Natural Science Foundation of China (No.21901191), the Fundamental Research Funds for the Central Universities and Wuhan University for financial support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.10.083.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction