
Computational insights into the effects of reagent structure and bases on nucleophilic monofluoromethylation of aldehydes
2022-06-20 07:59MengMengZhengHoDongTnYueqinSngXioSongXueJinPeiCheng
Chinese Chemical Letters 2022年5期
Meng-Meng Zheng, Ho-Dong Tn, Yueqin Sng, Xio-Song Xue,,*, Jin-Pei Cheng,c
a State Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China
b Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Acade my of Sciences, Shanghai 200032, China
c Center of Basic Molecular Science, Department of Chemistry, Tsinghua University, Beijing 100084, China
Keywords:Nucleophilic monofluoromethylation Fluoroalkylation reagent DFT calculations Thermodynamic effect Lone pair repulsion
ABSTRACT Although fluorobis(phenylsulfonyl)methane (FBSM) and its cyclic analog 2-fluoro-1,3-benzodithiole-1,1,3,3-tetraoxide (FBDT) possess similar physicochemical properties, Shibata et al. found that FBSM failed to undergo nucleophilic monofluoromethylation of aldehydes regardless of the reaction conditions attempted (using various organic and inorganic bases).However, it was later discovered by Hu et al. that the nucleophilic monofluoromethylation could be accomplished by employing lithium hexamethyldisilazide (LiHMDS) as a base.Herein, we present an in-depth computational investigation into the intriguing effects of reagent structure and bases on the nucleophilic monofluoromethylation of aldehydes.The computations reveal the 1,4-diazabicyclo[2.2.2]octane (DABCO) catalyzed nucleophilic monofluoromethylation of benzaldehyde with acyclic FBSM is a thermodynamically unfavorable process mainly due to the destabilizing O···O lone pair repulsions in FBSM product, whereas such repulsion could be largely avoided in FBDT product because of its constrained five-membered ring structure.Employing LiHMDS as a base can not only facilitate the nucleophilic monofluoromethylation via Li–O interactions but also render the monofluoromethylation of benzaldehyde with FBSM thermodynamically favored.
The increasing importance of the monofluoromethyl group in pharmaceuticals and agrochemicals as well as materials development has spurred tremendous efforts in the development of practical reagents for their incorporation into target molecules [1–17].Among various fluoromethylating reagents, fluorobis(phenylsulfonyl)methane (FBSM) [18,19] and its cyclic analog 2-fluoro-1,3-benzodithiole-1,1,3,3-tetraoxide (FBDT) [20] are popular nucleophilic reagents that have been widely used in monofluoromethylation reactions [21–29].Interestingly, while FBSM and FBDT possess similar acidity (pKaof FBSM:14.3vs.FBDT:13.5)[30] and their carbanions have comparable nucleophilicity (Mayr’s nucleophilicity parameter (N) of FBSM:17.46vs.FBDT:19.03)(Scheme 1a) [31], contrasting outcomes were observed in their reactions with aldehydes.In 2010, Shibataet al.found that the nucleophilic monofluoromethylation of aldehydes could proceed well with FBDT in the presence of 1,4-diazabicyclo[2.2.2]octane(DABCO) (Scheme 1b), but no desired product was observed when the reagent was switched from FBDT to FBSM (Scheme 1c)[20] .However, Huet al.subsequently demonstrated that employing an organometallic base lithium hexamethyldisilazide (LiHMDS) in combination with FBSM could enable the nucleophilic monofluoromethylation reaction of aldehydes at low temperatures(Scheme 1d) [32].
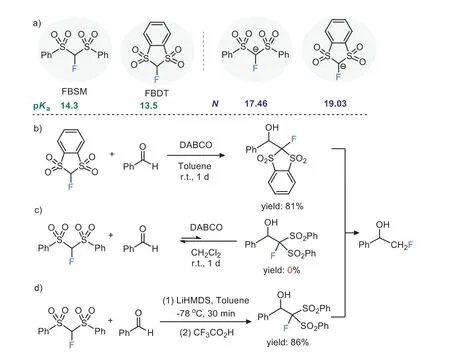
Scheme 1.(a) Physicochemical properties of FBSM and FBDT and (b-d) their applications in nucleophilic monofluoromethylation of benzaldehyde.
As a continuation of our research efforts on the structure-reactivity relationship and mechanism of fluorination/fluoroalkylation reagents [30,33-51], we report herein a computational study of these important nucleophilic monofluoromethylation reactions with an aim of understanding:(a) why the two monofluoroalkylating reagents FBDT and FBSM gave completely different results under identical conditions despite possessing similar physicochemical properties (Scheme 1bvs.1c)? (b) why changing the base from DABCO to LiHMDS could successfully promote the FBSM monofluoromethylation reaction(Scheme 1cvs.1d)?
Density functional theory (DFT) calculations were performed at the M06–2X/6–311+G(2d,p)-SMD//M06–2X/6–31G(d,p)-SMD level of theory (see Supporting information for computational details).The computed free energy profiles of nucleophilic monofluoromethylation of benzaldehyde with FBDT and FBSM are summarized in Fig.1.The first step is deprotonation of the reagent by DABCO to generate the hydrogen bond stabilizedαfluorocarbanion.The barrier for the deprotonation is slightly lower for FBDT (TS1a:8.7 kcal/mol) than for FBSM (TS1b:9.5 kcal/mol),in accord with our recent observation that the C–H bond is slightly more acidic in FBDT than in FBSM [30].Then, nucleophilic attack of the resultingα-fluorocarbanion (Int1) on benzaldehydeviaTS2,in which [DABCOH]+simultaneously interacts with both sulfone and carbonyl moietiesviahydrogen-bonding interactions, yields the products.The activation energy of nucleophilic addition for FBDT carbanion (12.6 kcal/mol) is slightly lower than that for FBSM carbanion (14.2 kcal/mol), in agreement with their nucleophilicity[31].To review the overall energy profiles, the nucleophilic addition is the rate-determining step.The overall free energy barrier for FBDT is only 1.9 kcal/mol lower than that for FBSM, suggesting the observed experimentally contrasting outcomes are less likely a result of the kinetic effect.On the other hand, the nucleophilic monofluoromethylation of benzaldehyde by FBSM is a thermodynamically unfavorable process (ΔG= 3.5 kcal/mol), whereas it is a slightly favorable process by FBDT (ΔG= -0.1 kcal/mol).Therefore, the distinct behavior between FBDT and FBSM under DABCO catalysis is primarily a result of the thermodynamic, rather than kinetic, effects.
Scrutiny of the structure of monofluoromethylation product Pbrevealed that the O···O distances between the carbonyl and sulfonyl oxygen are close to the sum of their van der Waals radii (Fig.2).Indeed, the computed electrostatic potential map indicates the destabilizing O···O lone pair repulsions in Pb.Such repulsions should be much weaker in Padue to the increased O···O distances as a result of the constraint of the five-membered ring in FBDT.Thus, the difference in the behavior of FBDT and FBSM can be mainly ascribed to the key difference in their structure (cyclicvs.acyclic) that results in different O···O lone pair repulsions in products.
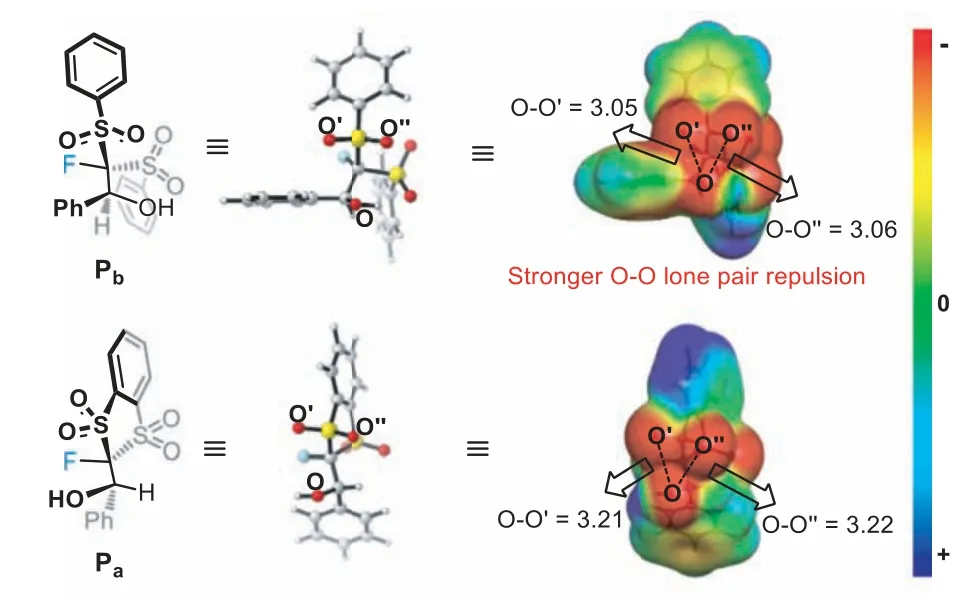
Fig.2.Calculated structures and electrostatic potential map of Pa and Pb.
Next, we explored why the nucleophilic addition of FBSM on benzaldehyde could be successfully promoted by changing the base from DABCO to LiHMDS.Computations of possible LiHMDS complexes suggest that the LiHMDS-benzaldehyde dimer is a stable complex that could work as a base (Scheme S1 in Supporting information for other higher energy LiHMDS complexes).As shown in Fig.3, the dissociation of LiHMDS-benzaldehyde dimer assisted by FBSM to form Int0cis endergonic by 5.2 kcal/mol.The deprotonation of FBSMviaTS1c, generating Li-coordinatedα-fluorocarbanion Int1c, is exergonic by 13.5 kcal/mol.The barrier for the LiHMDS-deprotonation is 7.8 kcal/mol lower than that for DABCO-deprotonation.The following nucleophilic attack ofαfluorocarbanion to C=O double bondviaTS2cleads to lithium carbinolate Int2cwith a barrier of only 4.4 kcal/mol, much lower than that of the DABCO-catalyzed process.This can be attributed to the greater stabilization of nucleophilic attack transition state by the Li–O interactions [32] as compared to hydrogen-bonding interactions.Then, the protonation of lithium carbinolate Int2cby CF3CO2H gives product Pb.The overall process is calculated to be exergonic by 27.2 kcal/mol, with a very low barrier of 7.8 kcal/mol(from Subbto TS1c).Clearly, the use of LiHMDS could not onlysignificantly reduce the barrier of nucleophilic monofluoromethylation (24.4DABCOvs.7.8LiHMDSkcal/mol), but also render the reaction to be a highly thermodynamically favorable process (+3.5DABCOvs.-27.2LiHMDSkcal/mol).This explains why the nucleophilic monofluoromethylation could be accomplished by employing lithium hexamethyldisilazide (LiHMDS) as the base.In fact, the organic base DABCO participates in the reaction as a catalyst (Scheme 2a), while the organometallic base LiHMDS serves as both reactant and activator (Scheme 2b).
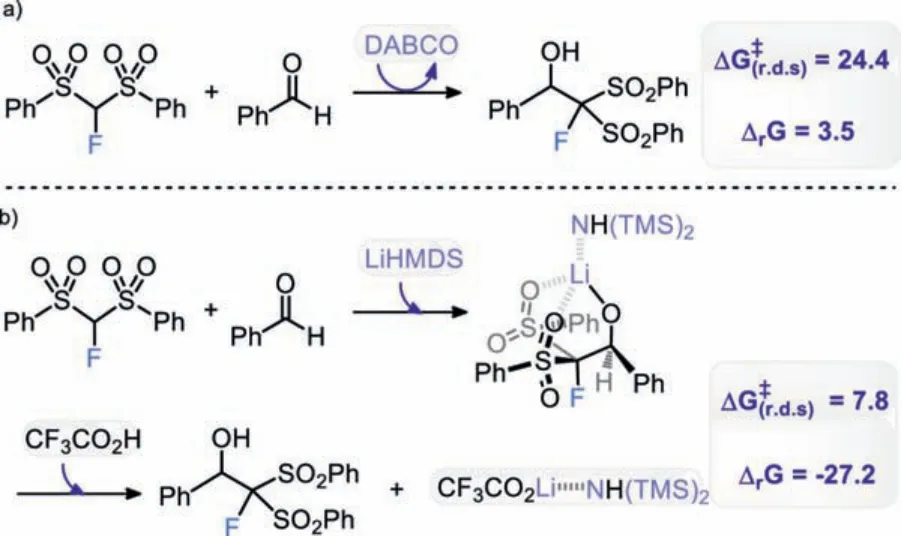
Scheme 2.Calculated Gibbs free energy (kcal/mol) changes of monofluoromethylation of benzaldehyde with FBSM by bases DABCO (a) or LiHMDS (b).
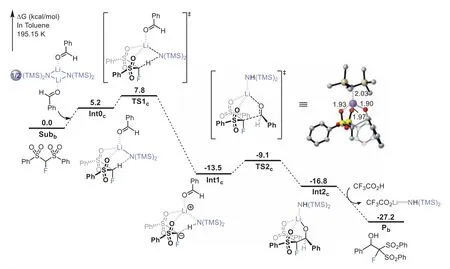
Fig.3.Calculated energy profile for monofluoromethylation of FBSM on benzaldehyde, using the LiHMDS as the base.
In summary, we have performed a computational study on the effects of reagent structure and bases on nucleophilic monofluoromethylation of aldehydes.The contrasting outcomes between cyclic FBDT and acyclic FBSM in DABCO-catalyzed nucleophilic monofluoromethylation of benzaldehyde are primarily a result of the thermodynamic effect.The nucleophilic monofluoromethylation of benzaldehyde with acyclic FBSM is a thermodynamically unfavorable process due to the O···O lone pair repulsions in the product.Such repulsions can be largely avoided in the FBDT product due to its cyclic structure.The use of LiHMDS can not only facilitate the nucleophilic monofluoromethylationviaLi–O interactions, but also render the nucleophilic monofluoromethylation reaction thermodynamically favored.The insights obtained regarding reagent structure and bases on nucleophilic monofluoromethylation should be valuable in future efforts towards the design of new reactions.
Declaration of competing interest
The authors declare no competing financial interest.
Acknowledgments
This work was supported by the Natural Science Foundation of China (Nos.22122104, 21933004 and 21702098).We thank professor Faguang Zhang at Tianjin University for helpful discussions.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.11.045.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction