
Recent advances in tertiary amine Lewis base-promoted cycloadditions of allenoates
2022-06-20 07:59MnmnSongJingZhoErQingLi
Chinese Chemical Letters 2022年5期
Mnmn Song, Jing Zho, Er-Qing Li,*
a College of Chemistry, Green Catalysis Center, Zhengzhou University, Zhengzhou 450001, China
b Xuchang Environmental Monitoring Center, Xuchang 461000, China
Keywords:Lewis base Tertiary amine Annualtion Allenoates Olefin
ABSTRACT Lewis base-catalyzed annulations of allenoates have been one of the most powerful synthetic strategies for the synthesis of various valuable cycles, especially in the preparation of biologically active natural products and pharmaceuticals.Generally, the effective Lewis bases mainly include tertiary phosphine,NHC and tertiary amine catalysts, among those catalysis, tertiary amine Lewis bases have proven to be effective catalysts for a range of synthetic transformations.In the past decades, tremendous progress involving tertiary amines-promoted cycloaddition of allenoates has been made in the chemoselective construction of valuable motifs.This review describes a comprehensive and updated summary of tertiary amine Lewis base-promoted annulation reactions of allenoates.Diverse reactivities, chemoselectivties and detailed reaction mechanisms will be highlighted in this review.
1.Introduction
Cycloadditions are among the most reliable bond-forming strategies in organic synthesis, and are widely used in synthesizing various valuable cycles in a single step, especially in the preparation of pharmaceuticals, bioactive natural products and agrochemicals [1,2].In the past several decades, synthetic chemists have made tremendous efforts to explore novel synthetic strategies and resolve the challenging problems in classic cycloadditions by using different catalytic systems involving metal catalysis, enzyme catalysis and organocatalysis.Among these fields, organocatalytic cycloaddition reaction, with its advantages of being environmentally friendly, having metal free residues, requiring simple and mild reaction conditions, has recently emerged as one of the most powerful catalytic strategies for the construction of carboand hetero-cycles.Lewis base catalysis, as a class of organocatalysis, occupies a preeminent position since the Rauhut–Currier reaction [3] and Morita–Baylis–Hillman reaction [4] are discovered in the 1960s.In particular, Lu and coworkers were the first to report the phosphine-catalyzed [3+2] cycloaddition reaction of activated alkenes in 1995 [5], and Lewis base catalysis has attracted interest from a large number of research groups.Many Lewis bases promoted cycloadditions of electro-deficient alkenes, allenoates,alkynes and MBH derivatives have been discovered in laboratories worldwide [6–16].
The conformations of tertiary amines are pyramidal and have a pair of nonbonding electron pairs, which represent the most commonly recognized form of Lewis base catalysts.With the development of Lewis base-catalyzed cycloaddition, tertiary amine catalysts have proven to be effective catalysts for a range of synthetic transformations.Usually, tertiary amines are generally more basic and less nucleophilic than similarly organophosphines, which show different catalytic reactivities and selectivities in the reactions.Thus a review focusing on amines as Lewis base catalysts in the reactions of electron-deficient olefins is highly desirable [17].
Allenoates, as a class of electron-deficient olefins, are attractive substrates for Lewis base catalysis due to their diverse reactivities.As illustrated in Scheme 1, nucleophilic addition of a tertiary amine catalyst to the electrophilicβ-carbon of allenoate results in the generation of a zwitterionic intermediate.The zwitterionic intermediate can be depicted in several ways, including anion localization at theα-carbon,γ-carbon or delocalized, as C1, C2 or C3 synthons (Scheme 1).Considering these resonance structures, it is easy to understand the versatile reactivities of allenoates in the presence of different electrophilic–coupling partners.
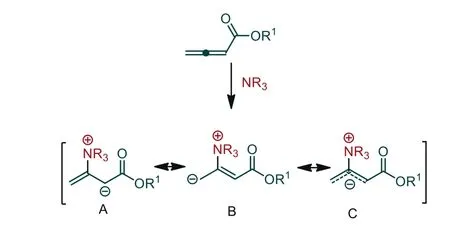
Scheme 1.Reactivity of allenoates in the presence of a tertiary amine.
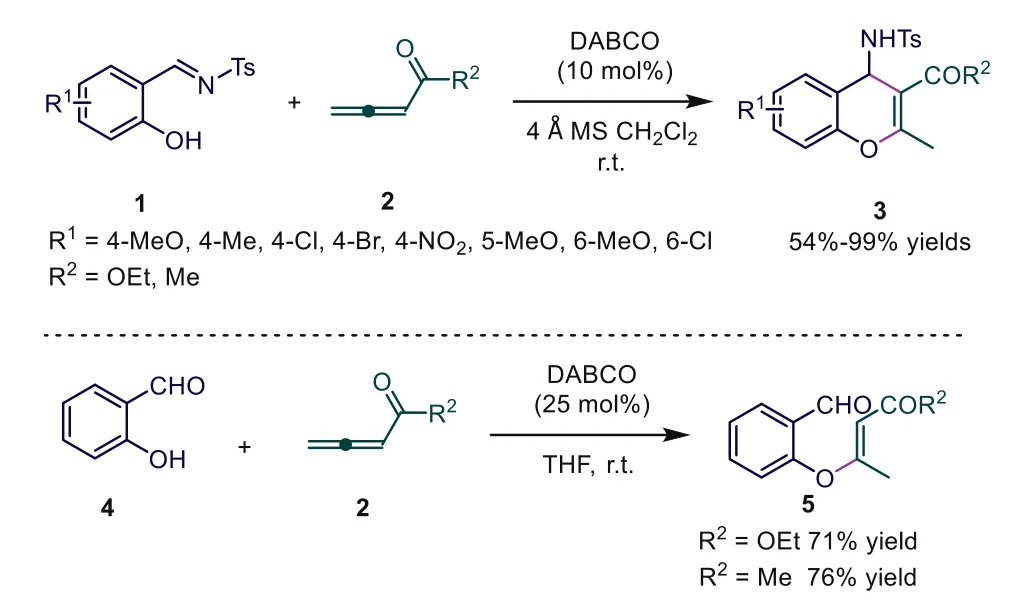
Scheme 2.DABCO-catalyzed [4+2] cycloaddition of allenoates/penta-3,4-dien-2-one with salicyl N-tosylimine.
In this review, we summarize recent progresses in tertiary amine Lewis base-promoted cycloadditions of allenoates.The diverse reactivity, various reaction modes and proposed mechanisms will be described in detail in the review.We hope that this tutorial review provides our readers with a systematic picture of this research field, leading to further development in the reporting of these new reaction modes.
2.Tertiary amine-catalyzed [4+2] cycloaddition of allenoates with electrophiles
As early as 1988, Tsuboi and coworkers first reported a DABCOcatalyzed Morita–Baylis–Hillman reaction of allenoates with aldehydes [18].DABCO-catalyzed [4+2] cycloaddition of allenoates was first discovered in 2005.In the literature, Shi and coworkers used ethyl 2,3-butadienoate/penta-3,4-dien-2-one and salicylN-tosylimines as reactive substrates to give highly functionalized chromene derivatives 3 in good to excellent yields [19].The authors found that molecular sieves of 4 Å as a desiccant could prevent the decomposition of imines by ambient moisture and improve the yield.Interestingly, allenoates were used as a novel C2 synthon in the [4+2] cycloaddition.However, the authors used salicylaldehyde 4 as a substrate instead of salicylN-tosylimines 1, and the corresponding Michael addition product 5 was formed rather than chromenes.The authors thought that the alkoxide adduct formed by the aldol reaction underwent proton transfer more slowly than the sulfonamide anion counterpart; and retroaldol reaction effectively competed to give the starting anion, and direct elimination of DABCO occurred to provide the product 5(Scheme 2).
The authors proposed a plausible reaction mechanism.As illustrated in Scheme 3, nucleophilic attack of the tertiary amine with the allenoate generated a zwitterionic intermediate 6, which deprotonated the phenol group in imine 1 to give intermediates 7 and 8.Next, a tandem Michael addition/ Mannich reaction, followed by proton transfer and elimination, produced chromene 3 and regenerated the catalyst.Accounting for the longer reaction time required for imines with electron-withdrawing groups on the benzene rings because of the decreased nucleophilicity of the oxygen atom, the authors thought that the Michael addition step might be the rate-determining step (Scheme 3).
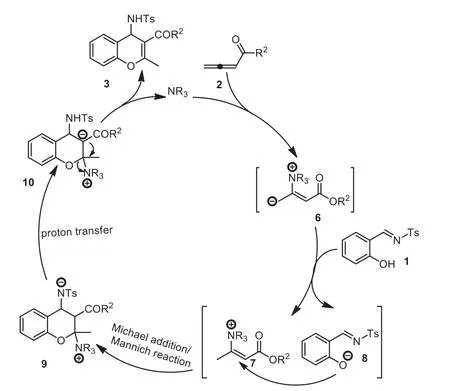
Scheme 3.Plausible reaction mechanism for DABCO-catalyzed [4+2] cycloaddition of salicyl N-tosylimines.
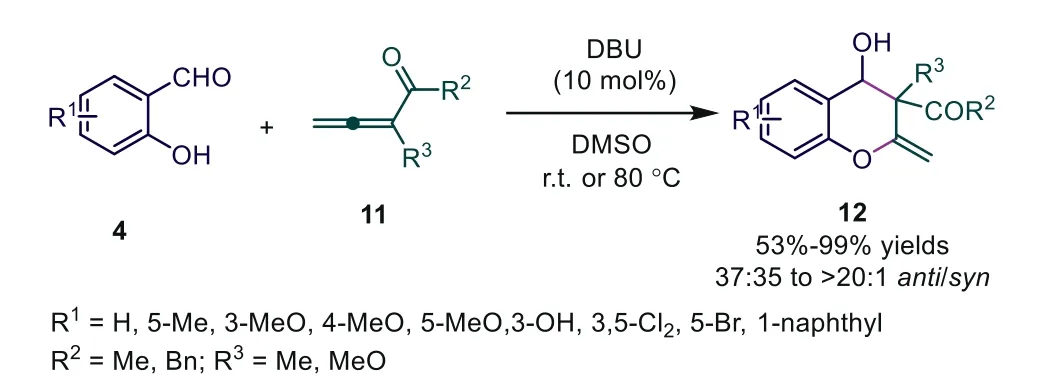
Scheme 4.DBU-catalyzed [4+2] cycloadditions of salicylaldehydes.
Based on previous work, Shi and coworkers wished to realize tertiary amine-catalyzed cycloadditions of salicylaldehydes with allenoates [20].By screening different allenoates, the authors found that 3-methylpenta-3,4-dien-2-one, 3-benzylpenta-3,4-dien-2-one, or ethyl 2-methylbuta-2,3-dienoate 11 with salicylaldehydes 4 were used in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene(DBU), and the [4+2] cycloaddition reaction proceeded smoothly,providing the corresponding functionalized 2H-1-chromenes 12 in good to excellent yields with moderate to good diastereoselectivities.This experiment suggested that the temperature played a very important role in the diastereoselectivity of this reaction.Only ananti-adduct was obtained when the temperature was increased to 80 °C, albeit with a slightly lower yield (81% yield).While the reaction occurred at 20 °C, 12 was obtained in 99% yield as a pair of diastereoisomers (anti/syn:74:25) (Scheme 4).
According to Shi’s report,αandβsites of allenoates participate in the formation of chemical bonds.Owing to the multiple reactive sites of allenoates, synthetic chemists have wanted to explore novel bonding strategies.In 2011, Ye and Wang reported novel DABCO-catalyzed [4+2] cycloadditions of allenoates with arylidenoxindoles, affording the corresponding dihydropyran-fused indoles 14 in good yields with exclusive regio- and diastereoselectivities [21].In this report, the novelβandγsites of allenoates participated in the formation of chemical bonds.Further study showed that the electrical properties of substituents on arylidenoxindoles had a significant influence on selectivity, and mixtures(desired adducts 14 and double bond migrated isomers 14′) were obtained in 67% and 65% yields, respectively, when strong electronwithdrawing groups (3-NO2, 2-NO2) were introduced.Almost simultaneously, Shi and coworkers reported the DMAP-catalyzed regioselective [4+2] cycloaddition of isatin-derivedα,β-unsaturated diesters with ethyl 2,3-butadienoate, affording the corresponding cyclic products 16 in good to excellent yields under mild conditions (Scheme 5) [22].
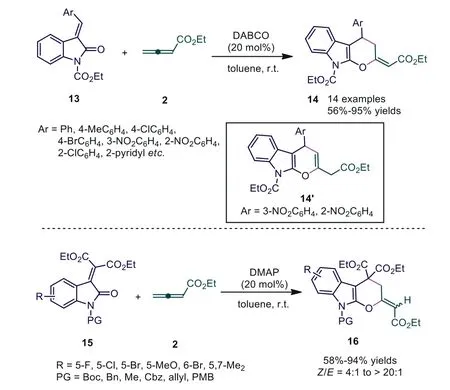
Scheme 5.DABCO or DMAP-catalyzed [4+2] cycloadditions of allenoates with oxindoles.
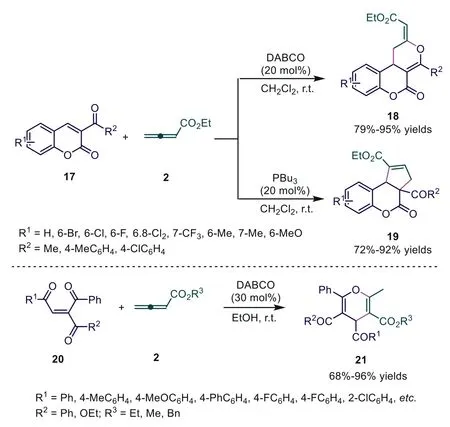
Scheme 6.DABCO or PBu3 catalyzed [4+2] or [3+2] cycloadditions of 3-acyl-2Hchromen-ones with ethyl 2,3-butadienoate.
In 2012, Shi and coworkers developed highly regioselective[4+2] and [3+2] cycloadditions of 3-acyl-2H-chromen-ones with ethyl 2,3-butadienoate catalyzed by DABCO and Bu3P, respectively, giving dihydropyran-fused and cyclopenten-fused chromen-2-one derivatives in moderate to excellent yields [23].Later,the authors applied 3-acyl(or alkoxycarbonyl)-1,4-enediones with 2,3-butadienoates as reactive substrates, finishing an efficient DABCO-catalyzed [4+2] cycloaddition of 3-acyl(oralkoxycarbonyl)-1,4-enediones with 2,3-butadienoates.In this report,αandβsites of allenoates participated in the formation of chemical bonds(Scheme 6) [24].
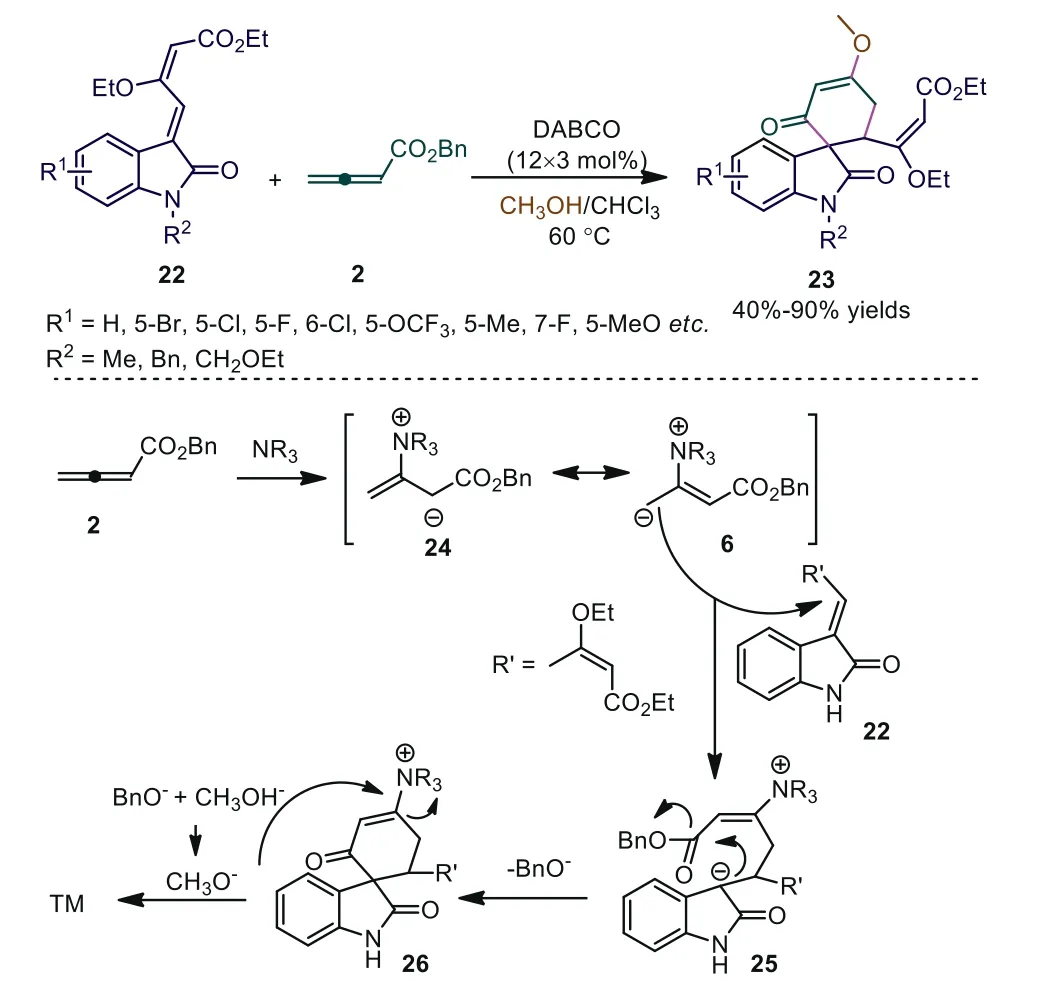
Scheme 7.DABCO-catalyzed [4+2] cycloadditions of methyleneoxindoles and allenoate.
Spirooxindoles are ubiquitous motifs of biologically active compounds [25–28].Because of their considerable medicinal potential,researchers have made efforts to develop efficient synthetic modes for the construction of spirooxindole scaffolds.In 2016, Meng and coworkers reported novel DABCO-catalyzed [4+2] cycloadditions of methyleneoxindoles and allenoate [29,30].The experiment results showed that 2,3-butadienoate first acted as a novel C4 synthon in a tertiary amine-catalyzed reaction, and the methoxy group acted as a nucleophilic reagent and appeared in the final product.The authors proposed a plausible mechanism for this domino reaction.First, acting as a nucleophilic trigger, DABCO attacked theβcarbon of the allenoate to produce intermediates 24 and 6.Subsequently Michael addition/intramolecular cyclization gave intermediate 26.Finally, the nucleophilic reagent (CH3O-) attacked 26 to give the desired product and regenerated DABCO (Scheme 7).
Catalyst-controlled regiodivergent cycloaddition has been a practical method to construct valuable cyclic skeletons in organocatalysis fields.In 2015, Swamy and co-workers reported divergent synthesis of functionalized dihydropyran derivatives and 1,1-alkyne (aldehyde)-substituted cyclopentenes by applying phosphine or amine Lewis base catalysts [31].When DABCO was used as the catalyst, [4+2] cycloaddition occurred, providing dihydropyran derivatives in 57%-86% yields.Meanwhile, [3+2] cycloaddition adducts were obtained in the presence of phosphines.It was noted that only enynals were used as reactive substrates, and phosphinecatalyzed [3+2] cycloaddition could proceed smoothly.The authors also attempted the reaction of allenoate 2a and enynal 27a by using simple chiral amines (20 mol%) in 1,4-dioxane as the solvent, and cycloaddition adduct 28a was obtained in 35% yield with 93%eewhen (DHQD)2PHAL was used as the chiral Lewis base catalyst (Scheme 8).
In 2017, Huang and coworkers reported a regiodivergent[4+2] cycloaddition ofα,β-unsaturated ketimines with 2,3-butadienoates, affording a series of hydropyridine derivatives in moderate to good yields under mild conditions [32–34].The authors found that when DABCO was used as a Lewis base catalyst,then theαandβsites of allenoates participated in the formation of chemical bonds, while theβandγsites of allenoates participated in the formation of chemical bonds in the presence of DMAP.Interestingly, absolute regioselectivity could be realized by only applying different nucleophilic tertiary amine catalysts (Scheme 9).
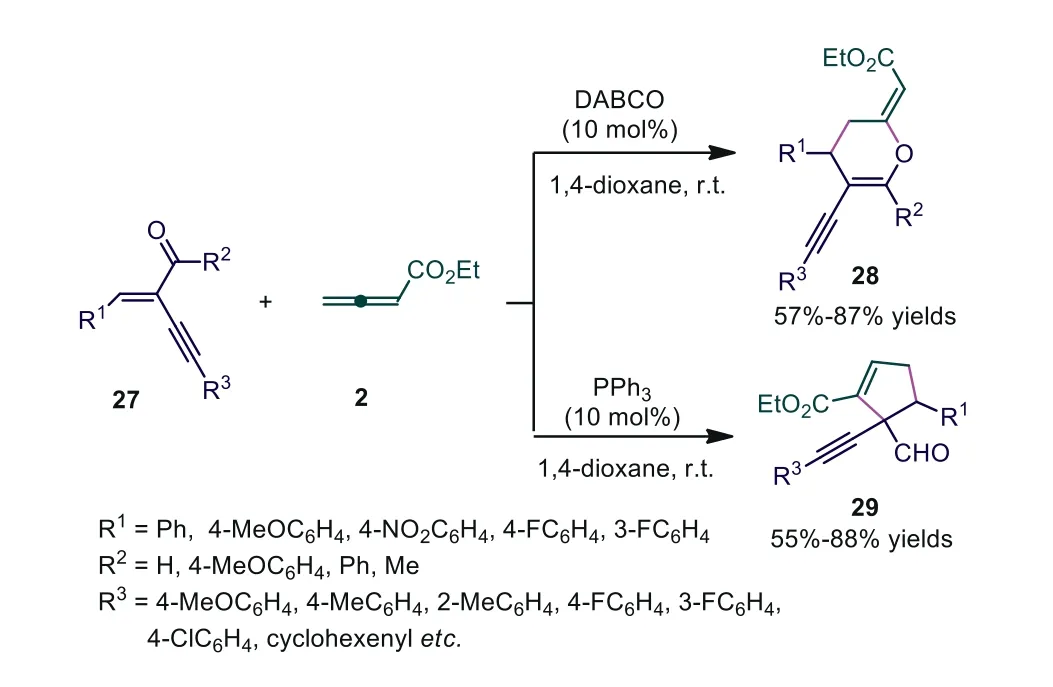
Scheme 8.Divergence in the reactivity between amine- and phosphine-catalyzed cycloaddition reactions of allenoate with enynals.
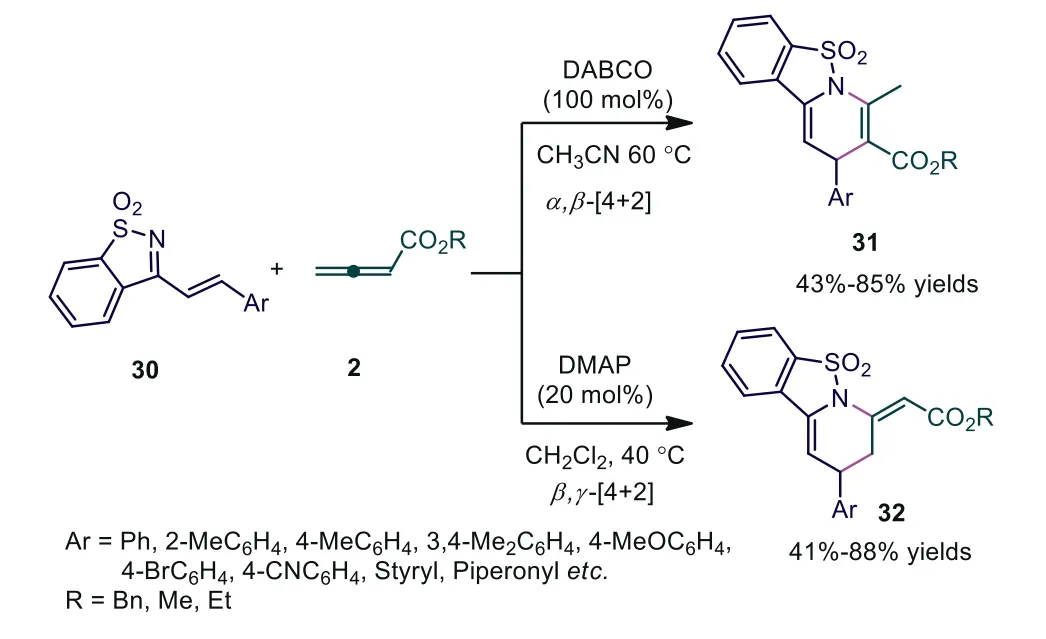
Scheme 9.Divergent synthesis of hydropyridine derivatives via nitrogen-containing Lewis base mediated regioselective [4+2] cyclizations.
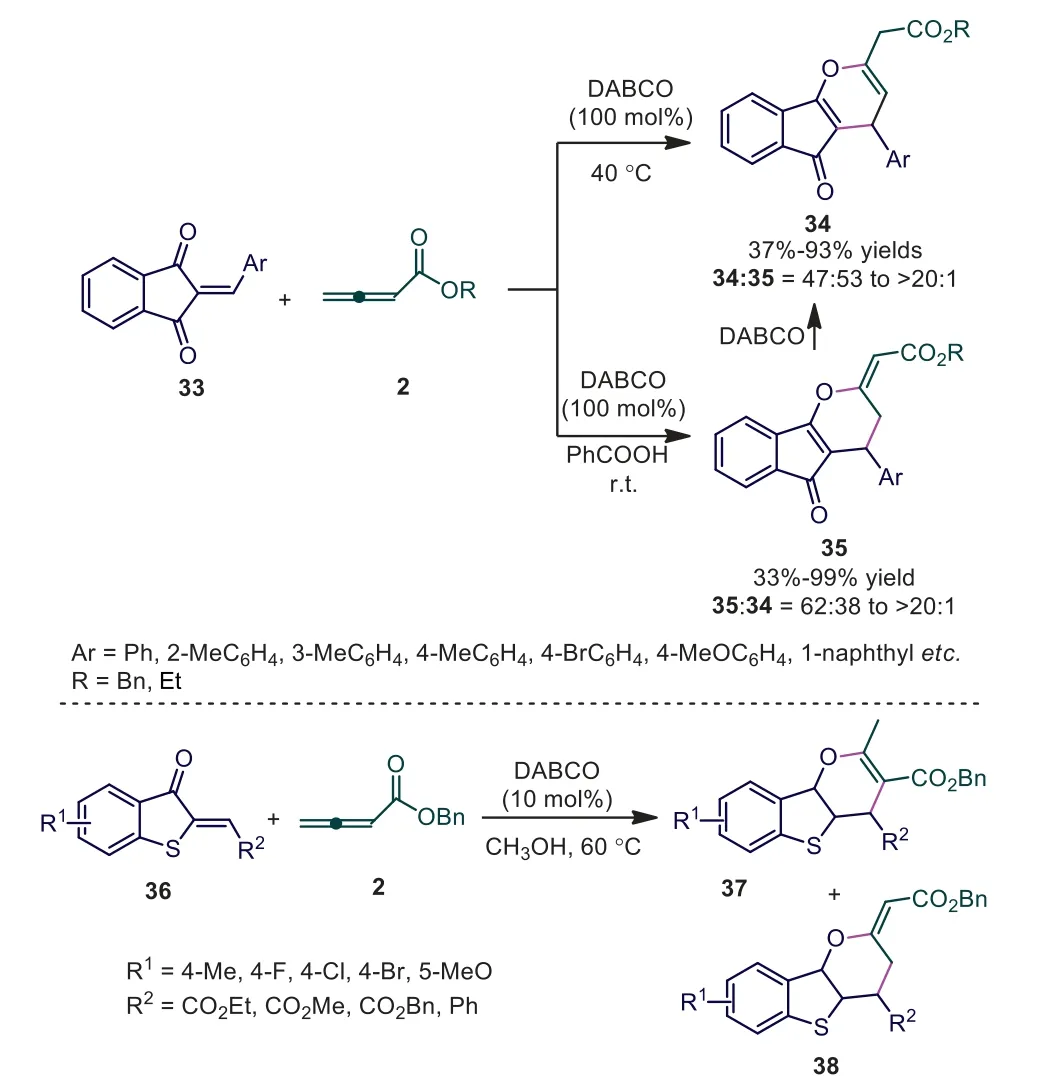
Our group is devoted to developing a novel Lewis basecatalyzed cycloaddition strategy [35–37].In 2018, we reported a DABCO-promoted [4+2] cycloaddition reaction of allenoates with 2-arylidene-1H-indene-1,3(2H)-diones, selectively giving annulated 4H-pyran 35 and annulated 3,4-dihydro-2H-pyran 34 in moderate to good yields, respectively [38].Further study suggested that the catalyst (DABCO) played a dual role in involving a Lewis basepromoted [4+2] cycloaddition and the subsequent Brønsted basemediated double bond isomerization.The role of the Brønsted base could be quenched selectively in the presence of a Brønsted acid(PhCO2H), and the desired annulated 3,4-dihydro-2H-pyran 35 was thus obtained as major product.Later, Meng and coworkers developed a DABCO-catalyzed [4+2] annulation reaction between 2-alkylidenebenzothiophene-3(2H)-ones and allenoates, providing a series of functionalized benzothiophene-fusedγ-pyran derivatives in moderate yields and selectivity (Scheme 10) [39].
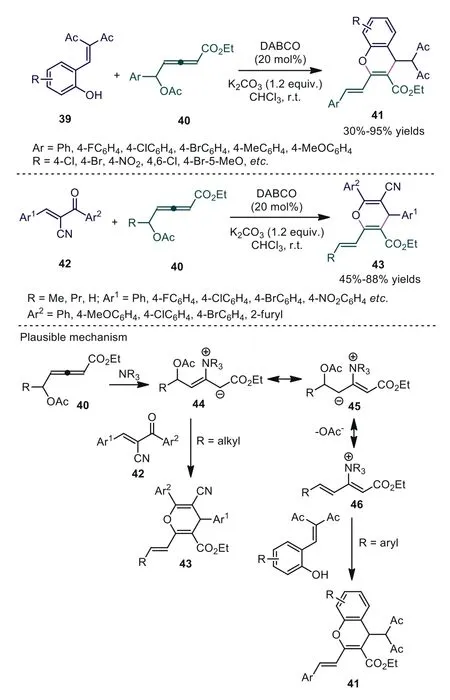
In early research,γ-substituted allenoates were thought to have low reactivity and selectivity and were not considered to be effective reactive substrates [40,41].In 2009, Huang and coworkers described novel phosphine-catalyzed [2+4] cycloadditions ofγmethyl allenoate in whichγ-CH3of the allenoates underwent cycloaddition to form chroman derivatives for the first time [42].Thus,γ-substituted allenoates have received considerable interest from synthetic chemists and continued with studies ofγsubstituted allenoates as synthons in cycloaddition reactions.In 2015, Tong and coworkers reported two different types of [4+2]cycloadditions ofδ-acetoxy allenoates in the presence of DABCO[43].The experiment suggested that the chemical behavior of allenoates under a DABCO catalyst was substrate dependent.Allenoates with an aromatic group atδCpreferentially reacted with salicylaldehydes, delivering the desired 4H-chromenes 41, while 4H-pyrans 43 could be obtained using allenoates with an alkyl group atδCand oxo dienes as reactive substrates.Mechanistically,the authors gave one reasonable explanation; for the case ofδ-aryl allenoates under tertiary amine catalysis, the reactivity of zwitterionic intermediate 45 might be reduced owing to steric hindrance.Thus, intermediate 45 might undergo 1,2-elimination of the acetoxy group, giving electrophilic intermediate 46.Whenδ-methylsubstituted allenoates were applied in this reaction, intermediate 45 might be unfavorable to formationviathe interaction of allenoates with DABCO due to the lack of a conjugation effect imposed by the aryl substituent.In this event, zwitterionic intermediate 44 would be dominant, which furnished a different [4+2]annulation with a suitable electrophile under tertiary amine catalysis (Scheme 11).
Scheme 10.DABCO-promoted [4+2] cycloaddition reaction of allenoates with 2-arylidene-1H-indene-1,3(2H)-diones.
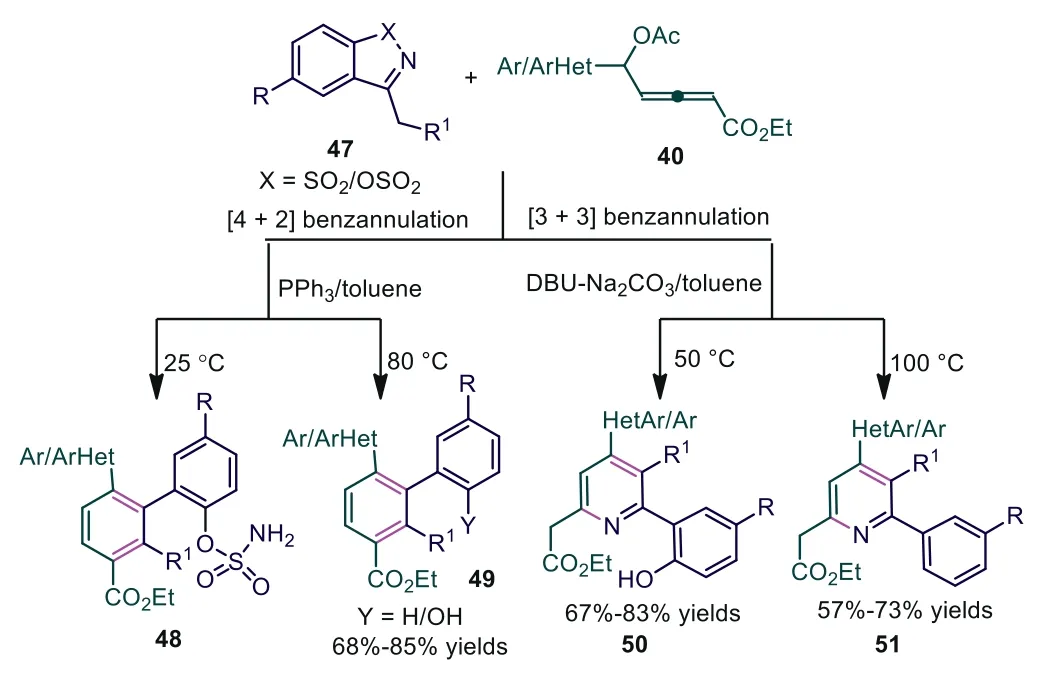
In 2020, Swamy and coworkers developed Lewis base-mediated[3+3] or [4+2] benzannulation reactions ofδ-acetoxy allenoates 40 with cyclicN-sulfonyl imines 47, giving teraryl motifs 48/49 or 2-pyridinyl acetates (α-pyridyl acetates) 50/51 by simply switching the Lewis bases [44].The authors found that changes in catalysts and temperature could result in the synthesis of different products.Under PPh3catalysis, the [4+2] benzannulation process gave functionalized teraryls by sequential Mannich coupling/C-N bond cleavage with retention or cleavage of the sulfamoyloxy group depending on the reaction temperature, while using DBU as the catalyst and Na2CO3as the additive, the [3+3] annulation involving sulfonyl eliminationviaO–S or C–S bond cleavage occurred, affording 2-pyridinyl acetates in good yields (Scheme 12).
More recently, Swamy and coworkers reported catalystdependent chemo-, regio-, and stereo-selective [4+2] cycloadditions involvingδ-acetoxy allenoate as a C4 synthon [45].The experiment showed that the nucleophilic distinction between DABCO and DMAP resulted in significant differences in reactivity and selectivity.When using DABCO as the catalyst, Na2CO3as the Brønsted base and AcOH as the Brønsted acid, the reaction occurred smoothly in toluene at 110 °C, affording desired adducts 52 in 55%–73% yields with absolute chemoselectivities.Benzannulation occurred in the presence of DMAP, giving unsymmetricalmteraryl scaffold 53 in high yield (78%).The mechanistic study revealed that spirocyclic 52 andm-teraryl 53 motifs were formed in entirely different pathways at later stages, and DABCO-mediated spiro-annulation occurredviaenol acetate-imine coupling.DMAPcatalyzed benzannulation proceededviaintramolecular Mannich coupling, proton transfers, and C-N bond cleavage (Scheme 13).
Scheme 11.DABCO-catalyzed divergent [4+2] annulations of δ-acetoxy allenoates with salicylaldehydes or oxo dienes.
Scheme 12.Lewis base-switched [3+3] and [4+2] benzannulation reactions of δacetoxy allenoates with cyclic N-sulfonyl imines.
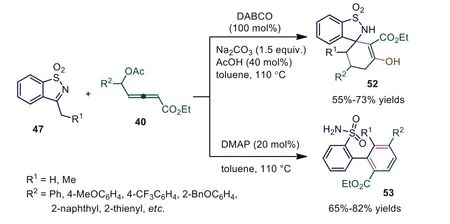
Scheme 13.Tertiary amine dependent chemo-, regio-, and stereoselective [4+2]annulations involving δ-acetoxy allenoate with cyclic N-sulfonyl imines.
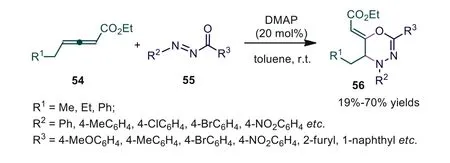
Scheme 14.DMAP-catalyzed [2+4] cycloaddition of allenoates and N-acyldiazenes.
N-Acyldiazenes are an important class of diazenes with distinctive reactivity and are commonly used in NHC-catalyzed cycloadditions with ketenes and aldehydes [46,47].In 2015, Meng and Wang reported an efficient DMAP-catalyzed [4+2] cycloaddition of allenoates andN-acyldiazenes, generating 1,3,4-oxadiazine derivatives in moderate to good yields [48].The experiment showed that the nucleophilicity of catalysts played a key role in the reactivity,and other amines (such as DABCO, Et3N, DBU) and phosphines (e.g.,PPh3, PBu3) shut down the cycloaddition completely (Scheme 14).
In 2016, Huang and coworkers reported DMAP-catalyzed [4+2]benzannulations involvingγ-substituted allenoates 54 and 1,3-bis(sulfonyl)butadienes 57 [49].The experiment suggested that only DMAP was used as the catalyst, and multisubstituted cyclohexene 58a and cyclohexadiene derivative 59a were produced in moderate yields.Some additives, including organic bases (e.g.,NEt3, DIPEA), inorganic bases (e.g., CH3CO2Na, NaOH, Cs2CO3), and Brønsted acid (PhCOOH), could tune the selectivity of the reaction to give polysubstituted arene 60a.It was noted that polysubstituted benzenes 60 with long alkyl chains, such as isobutyl ornpentyl, could also be obtained in good yields without undergoing rearrangement (Scheme 15).
In 2019, Miao and coworkers disclosed a tertiary aminecatalyzed regiodivergent [4+2] cycloaddition of allene ketones orα-methyl allene ketones with benzylidenepyrazolones, constructing the corresponding tetrahydropyrano [2,3-c] pyrazoles in moderate to good yields [50].Further study showed that formalα- andγ-[4+2] cycloadditions were found when quinine and DMAP were used as catalysts, respectively.The authors thought that the relatively electron-poor nucleophile quinine might stabilize carbon anion 24, leading to the thermodynamically favoredα-addition, while the relatively electron-rich nucleophile DMAP led to kinetically favoredγ-addition (Scheme 16).Later, Zhong and coworkers also realized [4+2] cycloaddition involving benzylidenepyrazolone with ethyl 4-phenylbuta-2,3-dienoate under DBU catalysis [51].
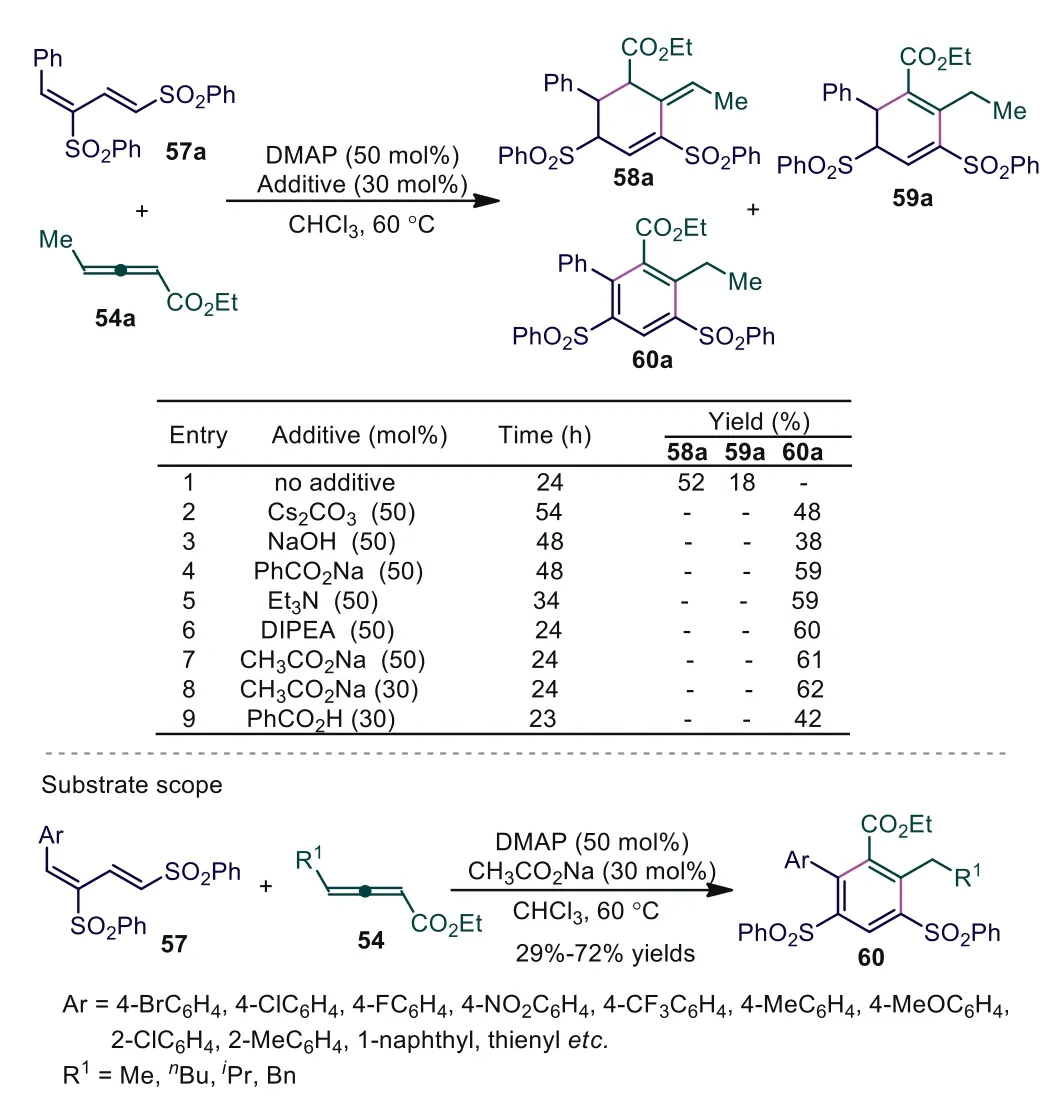
Scheme 15.DMAP-catalyzed [4+2] benzannulation of γ-substituted allenoates and 1,3-bis(sulfonyl)butadienes.
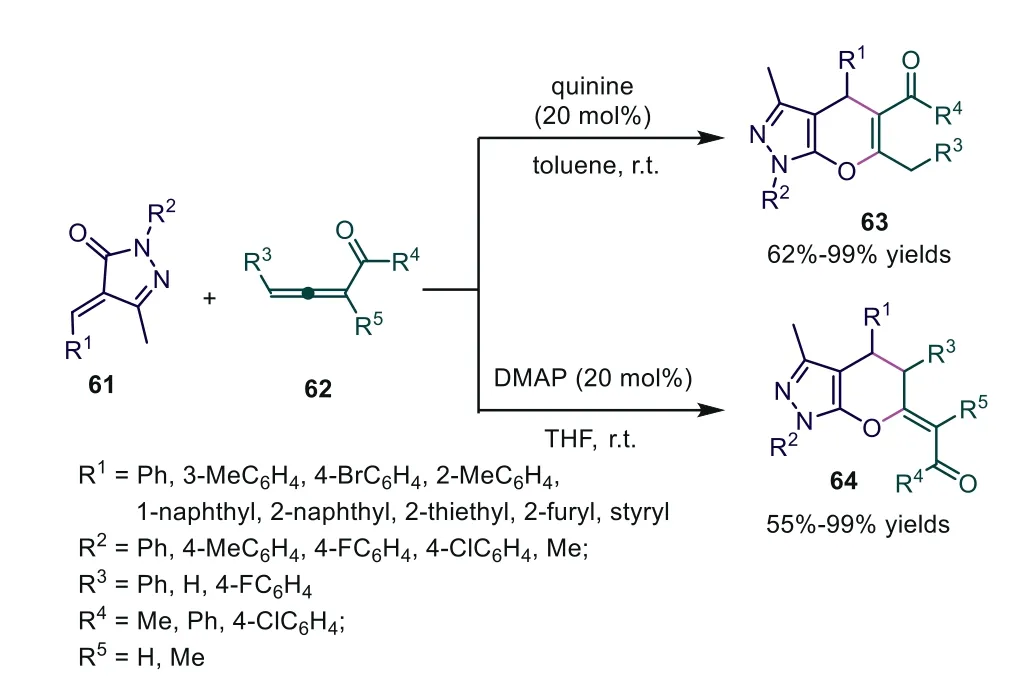
Scheme 16.Tertiary amine-catalyzed regiodivergent [4+2] cycloaddition of allene ketones or α-methyl allene ketones with benzylidenepyrazolones.
3.Tertiary amine-catalyzed [2+2] cycloaddition of allenoates with electrophiles
Azetidines are present as a partial structure of numerous natural products, biologically active compounds and pharmaceuticals.Thus, the development of effective methods for the assembly of functionalized azetidines from readily available starting materials is still of interest.In 2003, Shi and workers reported a tertiary amine-catalyzed cycloaddition reaction of ethyl 2,3-butadienoate or penta-3,4-dien-2-one withN-tosylated imines[52].Screening the Lewis bases, the authors found that [2+2]cycloaddition adducts were obtained in the presence of DABCO.When DMAP was used as a Lewis base catalyst, the reaction occurred smoothly by “abnormal” [3+2+1] cycloaddition, affording the desired products in 30%–60% yields.Interestingly, when a relatively stable aliphaticN-tosylated imine was applied under DMAP catalysis, the reaction still proceeded smoothly, giving the corresponding 3-[(2-methylpropenyl)(toluene-4-sulfonyl)-amino]-but-3-enoic acid ethyl ester 69 in a 20% yield (Scheme 17).
The authors proposed a reasonable mechanism, as shown in Scheme 18.First, DABCO and DMAP acted as nucleophilic triggers and gave zwitterionic intermediate (allylic carbanion) 6, which existed with resonance-stabilized intermediate 24 (enolate).When DABCO was used as a Lewis base catalyst, allylic carbanion 6 attacked theN-tosylated imine to produce intermediate 70, which underwent intramolecular cyclization to give intermediate 71.The final elimination of NR3from 71 afforded producted 66 and regenerated DABCO.Under DMAP catalysis, intermediate 24 was favorable, attacking anN-tosylated imine to give intermediate 72, which attacked anotherN-tosylated imine to give intermediate 73.Subsequent proton shift/intramolecular Michael addition gave intermediate 75.Finally, another proton shift/NHT elimination furnished product 67 and regenerated DMAP (Scheme 18).
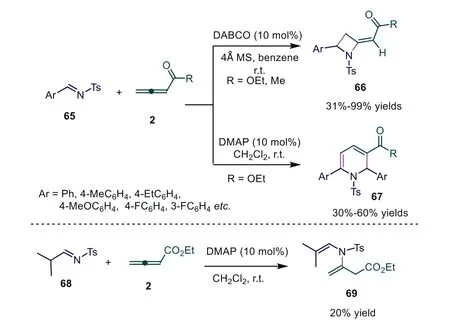
Scheme 17.Tertiary amine-catalyzed cycloaddition reaction of ethyl 2,3-butadienoate or penta-3,4-dien-2-one with N-tosylated imines.
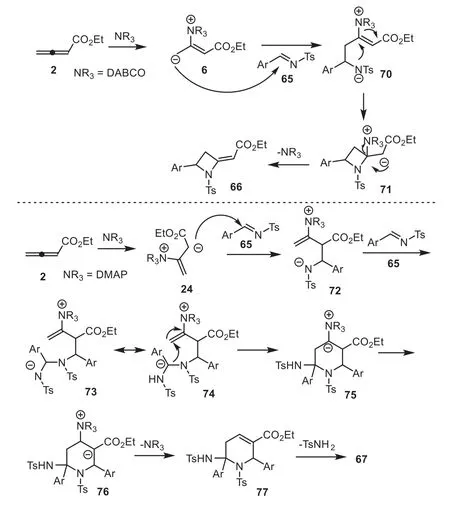
Scheme 18.Proposed mechanism for tertiary amine-catalyzed cycloaddition of allenoates with N-tosylated imines.
In 2011, Ye and coworkers discovered the DABCO-catalyzed[2+2] cycloaddition of allenoates and trifluoromethylketones, producing the corresponding 2-alkyleneoxetanes 79 in good yields with good diastereoselectivities [53].It was noted that only [2+2]cycloaddition adducts were obtained in the presence of either DABCO or DMAP compared to that in Shi’s report (Scheme 19)[52].In 2013, Selig and coworkers used bicyclic guanidine 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a tertiary amine Lewis base catalyst, also finishing the [2+2] cycloaddition of allenoates with trifluoromethylketones [54].
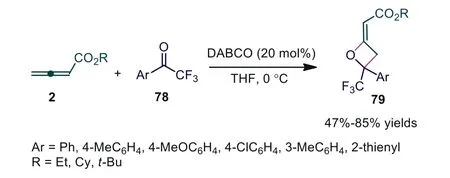
Scheme 19.DABCO-catalyzed [2+2] cycloaddition of allenoates and trifluoromethylketones.
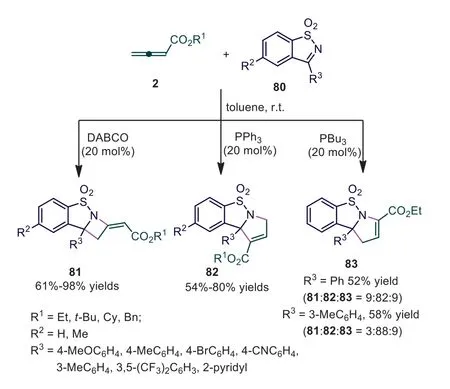
Scheme 20.Catalysts-controlled [2+2] or [3+2] cycloaddition of cyclic sulfonamides with allenoates.
Cyclic ketimines or their derivatives have important applications in organic synthesis and medicinal chemistry because of their biological activity [55,56].In 2012, Ye and coworkers reported a catalyst-controlled [2+2] orα-[3+2] cycloaddition of cyclic sulfonamides with allenoates, giving the corresponding sultam-fused azetidines and dihydropyrroles in good yields with high regioselectivities [57].Interestingly, [2+2] cycloadducts 81 and [3+2]cycloadduct 82 were formed exclusively when DABCO and PPh3were applied as the catalysts, respectively.The reaction catalyzed by PBu3gaveγ-[3+2] cycloadduct 83 predominately with litter cycloadducts 81 and 82 (Scheme 20).
In 2015, Xu and coworkers used azodicarboxylates as reactive substrates, finishing the DABCO-catalyzed [2+2] cycloaddition of allenoates [58].In this report, the authors realized formal [3+2]annulation of allenoates and azodicarboxylates for the formation of pyrazoline by a DABCO/PPh3relay catalytic strategy (Scheme 21).
In 2016, Silvani and coworkers used chiraltert-butanesulfinyl ketimines as electrophiles and achieved DABCO-catalyzed [2+2]cycloaddition of allenoates, producing enantiopure spirooxindolefused 4-methyleneazetidines in moderate to good yields [59].Further study showed thatα-substituted allenoates proved to be completely uncreative under the reaction conditions, and the authors thought that the result could be ascribed both to the stereoelectronic influence of the methyl group on the allenoate and to the high steric challenge inherent in the formation of the tetrasubstituted double bond joined to the spiroazetidine ring.More recently,Huang and Cheng described the DABCO-catalyzed [2+2] cycloaddition of quinone imine ketals with allenoates (Scheme 22) [60].
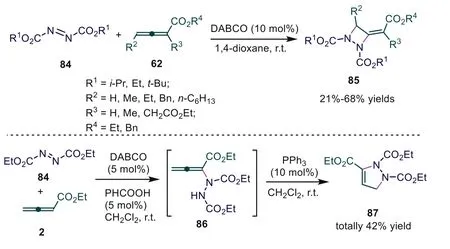
Scheme 21.DABCO-catalyzed [2+2] annulation of allenoates with azodicarboxylates.
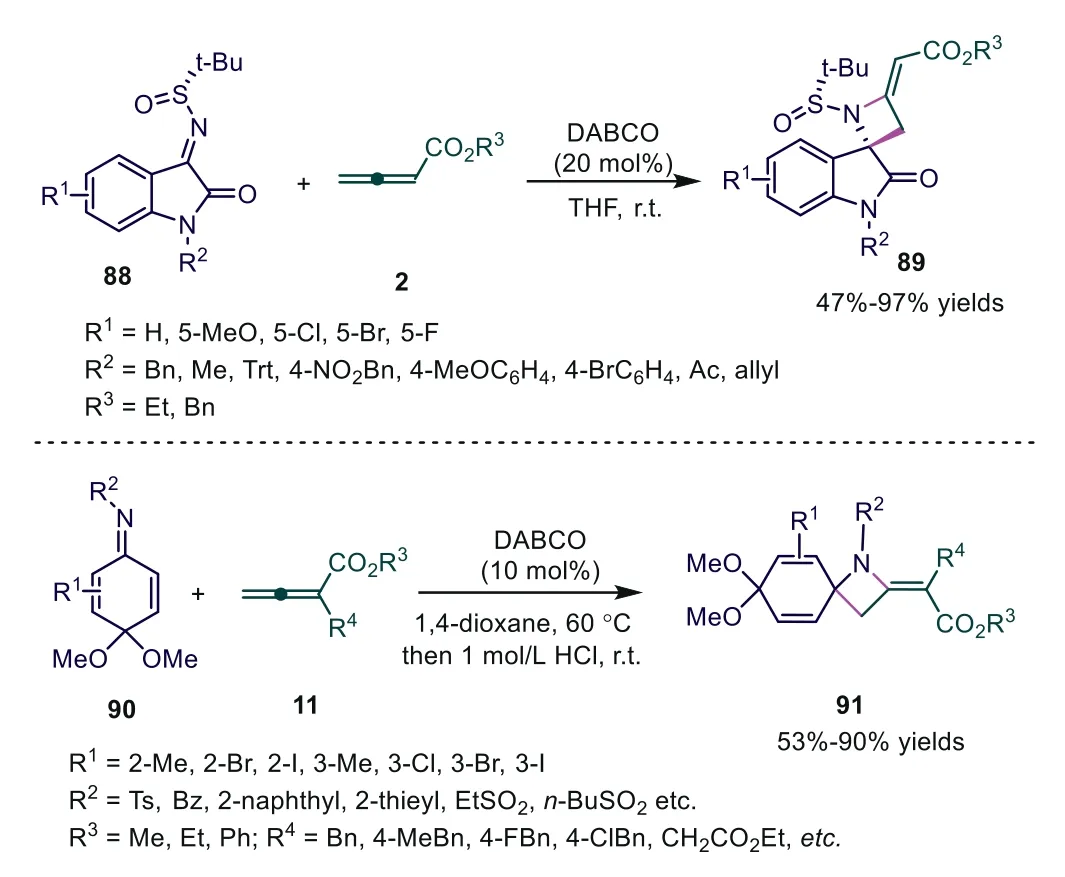
Scheme 22.DABCO-catalyzed [2 + 2] cycloaddition of allenoates with tertbutanesulfinyl ketimines or quinone imine ketals.
4.Tertiary amine-catalyzed [3+3] cycloaddition of allenoates with electrophiles
In 2003, Kwon and coworkers developed a novel strategy usingα-substituted allenoates as substrates to first achieve phosphinecatalyzed [4+2] cycloaddition [61].Inspired by this result, Tong and coworkers usedβ′-acetoxy allenoates as substrates and successfully applied them to phosphine-catalyzed [4+1] annulations with 3-oxo-3-phenylpropanenitrile.Based on this, the novel DABCO-catalyzed [3+3] cycloaddition ofβ′-acetoxy allenoates with 3-oxo-3-phenylpropanenitrile was developed, producing the corresponding 4-substituted 4H-pyrans 94 in 48%–99% yields [62].In a preliminary attempt, asymmetric [4+2] annulation was examined with quinine catalysts, affording 94a in 82% yield and 19%ee.The authors thought that this DABCO-catalyzed [3+3] annulation probably proceeded through a mechanism involving the destruction and subsequent reconstruction of the stereogenic center of theβ′-carbon (Scheme 23).
A plausible mechanism was proposed by the authors.As shown in Scheme 24, attack by DABCO to theβ-carbon of the allenoate initiated the addition–elimination process to yield intermediate 96.According to previous reports, it was unlikely that Michael-type addition of 93 to theγ-position of intermediate 96 was due to the poorer ability of the ammonium ion to stabilize the ylide.Apparently, intermediate 97 was generated by a Michael-type addition of 93 to theβ′-position of 96 with the help of a base.Next, 1,2-elimination of DABCO delivered intermediate 98, which was followed by 6-endo-trigtype oxo-Michael addition to form intermediate 100.Finally, compound 94 could be obtained by continuous isomerization and protonation.It was noted that intermediate 98 could be isolated when the reaction of 92 and 93 was conducted in benzene solvent for a short period of time, and the conversion of 98 to 94 was achieved with a longer reaction time (Scheme 24).
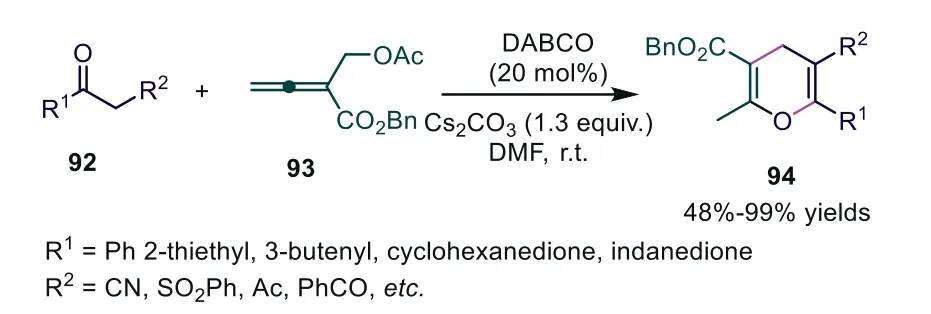
Scheme 23.DABCO-catalyzed [3+3] cycloaddition of β′-acetoxy allenoates with 3-oxo-3-phenylpropanenitrile.
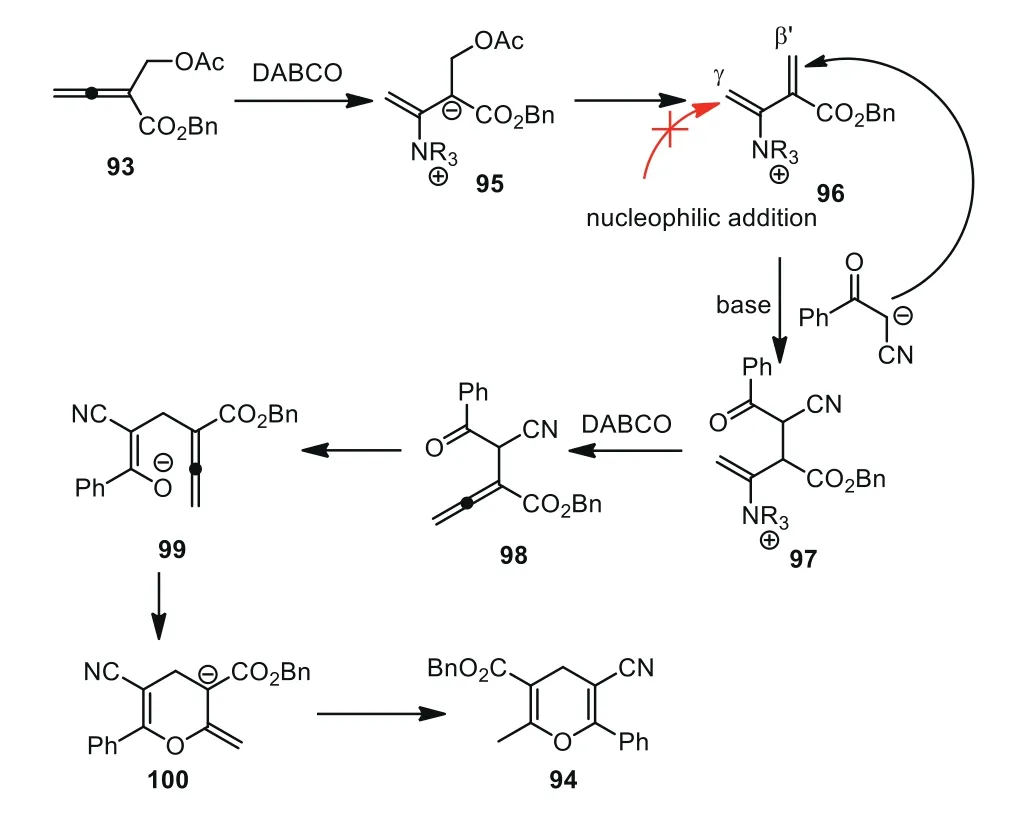
Scheme 24.Plausible mechanism for DABCO-catalyzed [3+3] cycloaddition of β′-acetoxy allenoates with 3-oxo-3-phenylpropanenitrile.
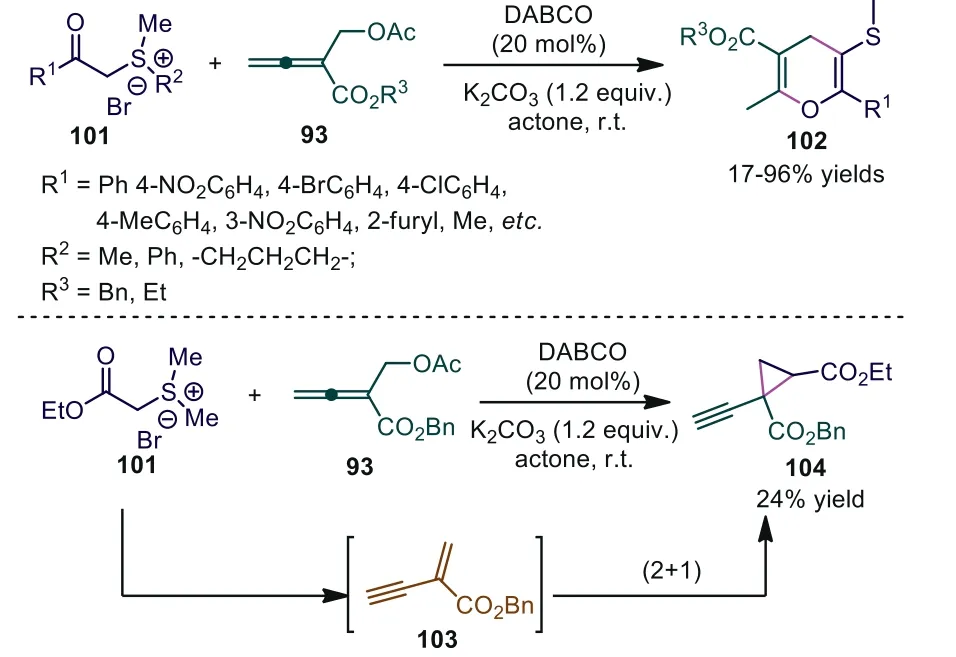
Sulfur ylides show unique and important reactivity in organic synthesis [63,64].Over the past decade, chemical transformations involving sulfur ylides have attracted much research interest, with a number of novel sulfur ylide-based reactions with high synthetic potentials having emerged for the synthesis of epoxide, aziridine and cyclopropane.In 2012, Tong and coworkers reported a DABCOcatalyzed [3+3] annulation betweenβ′-acetoxy allenoates and sulfur ylides, and they synthesized S-containing 4H-pyran 102 in good yields [65].Further study showed that only a 17% yield of 102j was obtained using methyl ketone-stabilized sulfonium salt as the nucleophile even with a prolonged reaction time.In addition, cyclopropane derivative 104 was obtained in 24% yield when ester-stabilized sulfur ylides were used under the same conditions.According to Lee’s report [66], the authors thought that the stability of key intermediate 103 played a critical role in the result(Scheme 25).
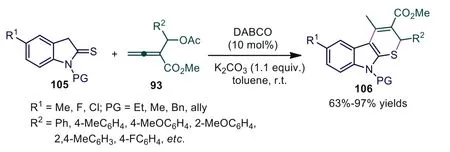
Later, the authors also achieved DABCO-catalyzed formal [3+3]annulations ofβ′-acetoxy allenoates with indoline-2-thiones, providing the desired thiopyrano[2,3-b]indoles 106 in 63%–97% yields(Scheme 26) [67].
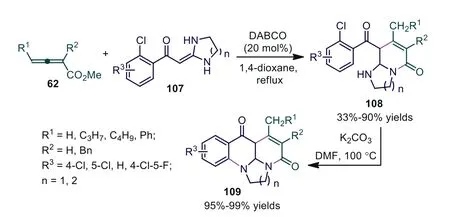
In 2011, Li and Wen reported a DABCO-catalyzed [3+3] cycloaddition of 2-(2-chloroaroyl)methyleneimidazolidines with allenoates, affording functionalized imidazo(pyrido)[1,2-a]pyridines 108 in moderate to good yields [68].Further study revealed that the corresponding imidazo(pyrido)[3,2,1-ij][1,8]naphthyridines 109 were obtained in the presence of K2CO3(1.0 equiv.) in DMF at 100 °C for approximately 6 h in almost quantitative yields of 95%–99%.The experimental results suggested that this domino process involved nine reactive sites.One C-C bond, two C-N bonds, and two new rings are constructed with all reactants efficiently utilized in the chemical transformation (Scheme 27).
Scheme 25.DABCO-catalyzed [3+3] annulation between β′-acetoxy allenoates and sulfur ylides.
Scheme 26.DABCO-catalyzed formal [3+3] annulations of β’-acetoxy allenoates with indoline-2-thiones.
Scheme 27.DABCO-catalyzed [3+3] cycloaddition of 2-(2-chloroaroyl)methyleneimidazolidines with allenoates.
5.Other tertiary amine-catalyzed cycloadditions of allenoates with electrophiles
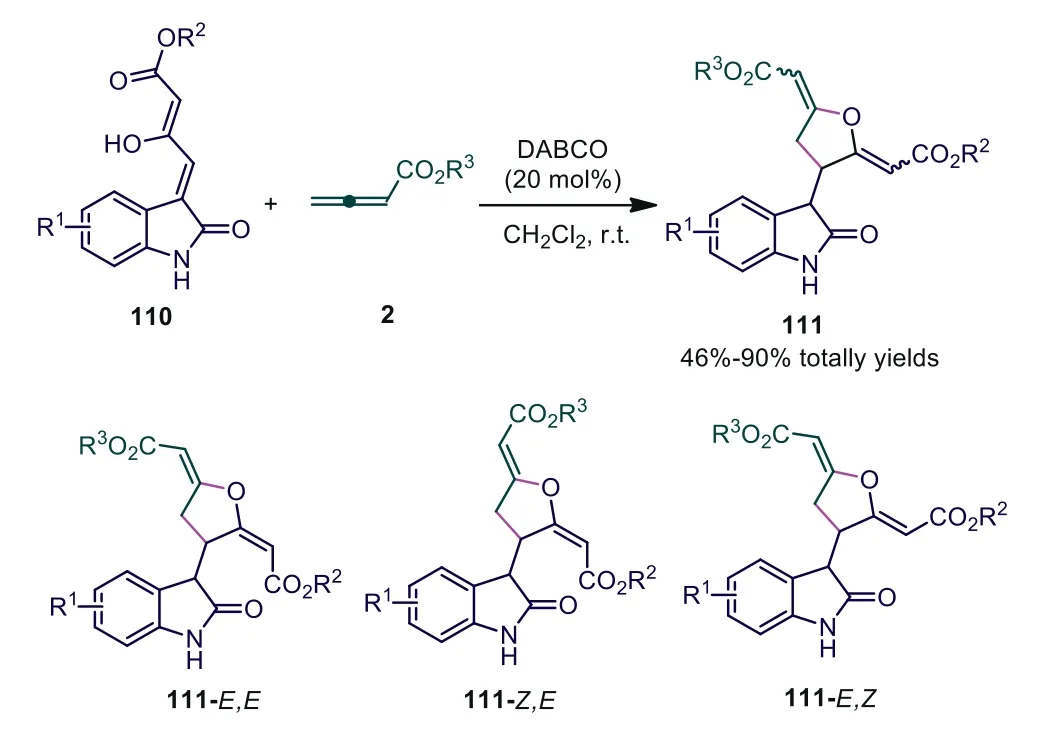
In 2014, Meng and coworkers reported the DABCO-catalyzed[3+2] cycloaddition reaction between 3-oxo-4-(2-oxoindolin-3-ylidene) butanoates and allenoates, furnishing 2,3,5-substituted tetrahydrofurans in high yield [69].In this report, moderate chemoselectivity was obtained in most cases, and three isomers were simultaneously synthesized in which two of them could be isolated.Interestingly, isatins withoutN-protecting groups were used to successfully finish the cycloaddition in the reaction, which was highly desirable for practical synthetic applications (Scheme 28).
More recently, Ma and coworkers reported the Et3N-catalyzed[3+2] cycloaddition reaction of allenoates with diazoesters, diazoketones, and diazoamides, leading to a group of pyrazoles in 56–90% yields (Scheme 29) [70].
Scheme 28.DABCO catalyzed the [3+2] cycloaddition reaction between 3-oxo-4-(2-oxoindolin-3-ylidene)butanoates and allenoates.
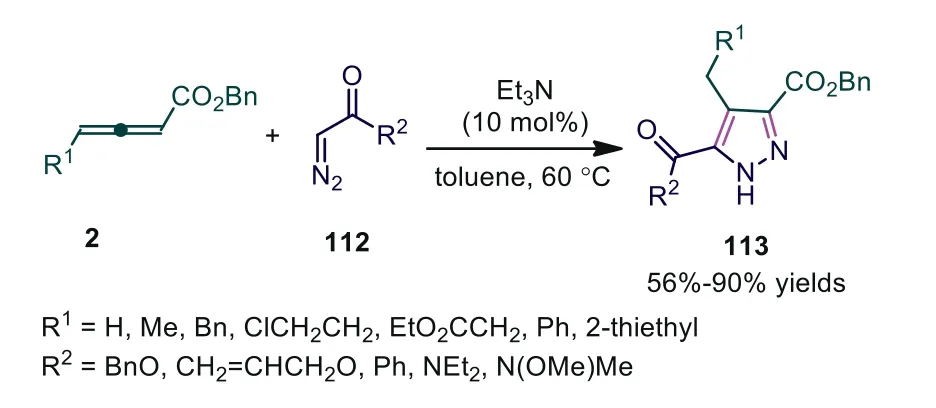
Scheme 29.Et3N-catalyzed [3 + 2] cycloaddition reaction of allenoates with diazo compounds.
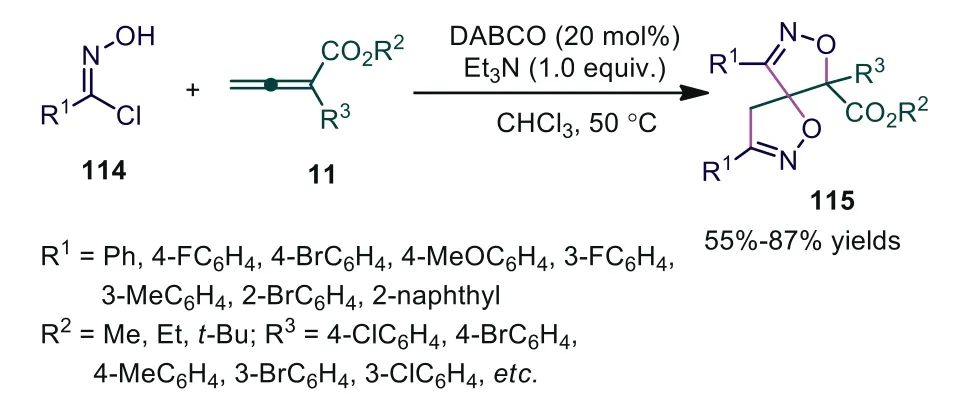
Scheme 30.DABCO-catalyzed double [3 + 2]-cycloadditions between nitrile oxides and allenoates.
Allenoates have proven to function as 1,2-, 1,3-, and 1,4-dipole synthons when reacting with a variety of electrophilic partners in the presence of tertiary amines.Only two chemical bonds are generated in most reactions.Because of the multiple reactive sites in allenoates, it was possible that more sites took part in the formation of chemical bonds at the same time [71].In 2018, Li and coworkers discovered double [3+2] cycloadditions between nitrile oxides and allenoates in the presence of DABCO with Et3N in which DABCO was used as a nucleophilic Lewis base catalyst with Et3N as the Brønsted base [72].Theα,β,γsites of allenoates took part in the formation of chemical bonds in the reaction.It was noted that nitrile oxides with aliphatic substituents were not suitable substrates in this reaction (Scheme 30).
In 2020, Huang and coworkers reported the DMAP-catalyzed[4+1]/[3+3] domino sequential cycloaddition reaction betweeno-aminotrifluoroacetophenones andβ′-acetoxy allenoates, which afforded a series of CF3-containing tetrahydropyrano[3,2-b]indole products in high to excellent yields with a single diastereoselectivity [73].In previous reports, all nucleophiles were added to theβ′-position of intermediate 96 because no empty orbital in the ammonium ion of intermediate 96 stabilized the carbanion in the subsequently generated ammonium ylide.To overcome these limitations, the authors used DMAP with aπ-conjugated system as the catalyst, and the p-πconjugation between the carbanion and the aromatic ring of DMAP could stabilize the corresponding ammonium ylide.Thus, Huang first realized the addition of nucleophiles to theγposition of intermediate 96 (Scheme 31).
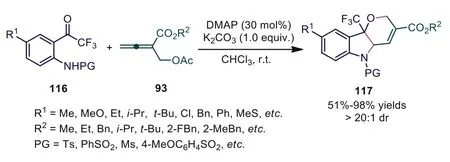
Scheme 31.DMAP-catalyzed [4+1]/[3+3] domino sequential cycloaddition reaction between o-aminotrifluoroacetophenones and β′-acetoxy allenoates.
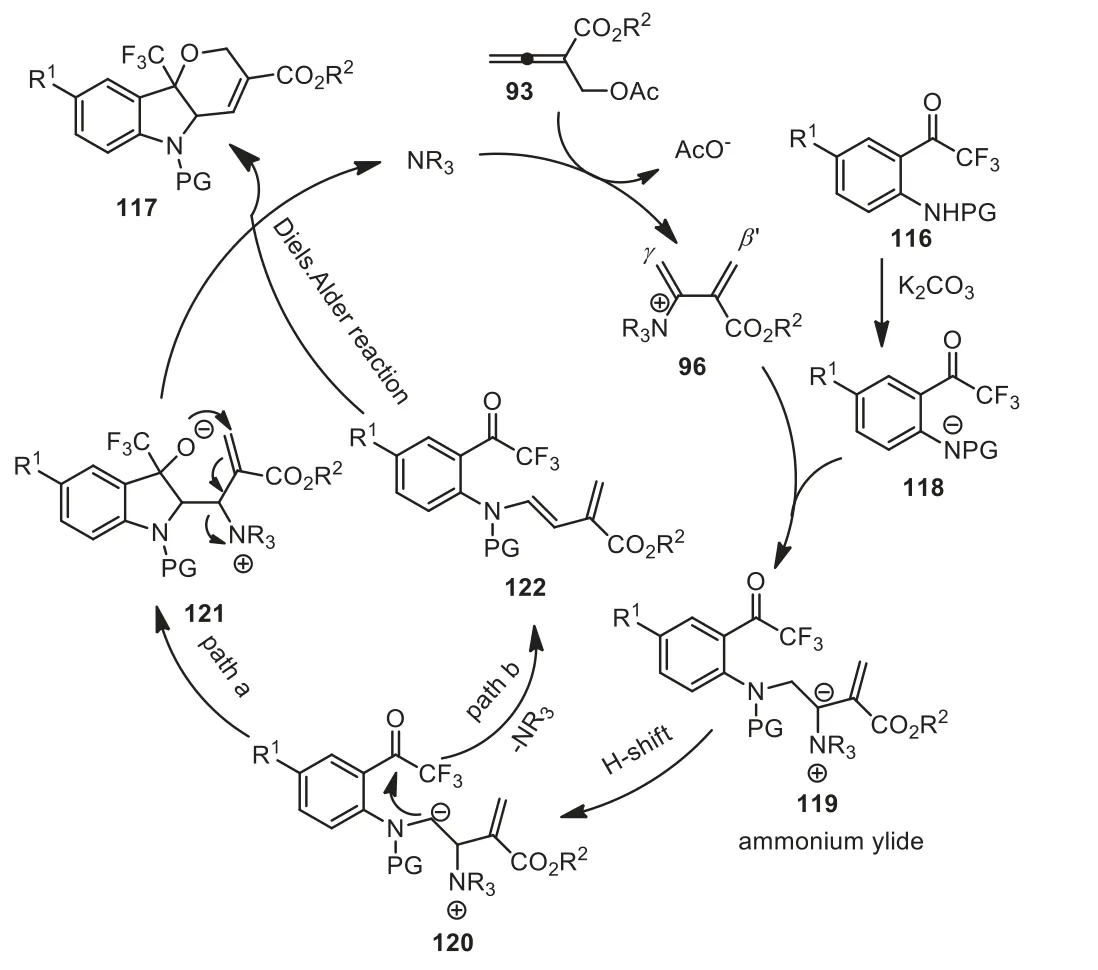
Scheme 32.A plausible reaction pathway of DMAP-catalyzed [4+1]/[3+3] domino sequential annulation of allenoates.
The author proposed a plausible reaction pathway, as showed in Scheme 32.Nucleophilic addition of DMAP to allenoates was followed by 1,2-elimination of acetate to generate intermediate 96.Subsequently,γ-addition of intermediate 118 to intermediate 96 formed intermediate 119.Next, tandem proton shift (120) /intramolecular 1,2-addition/intramolecular SN2′ substitution in intermediate 121 yielded product 117 with DMAP catalyst regeneration.The authors thought that it was also possible for it to undergo tandem 1,2-eliminationviathe DMAP/intramolecular Diels-Alder reaction, affording product 117 directly (Scheme 32).
Lewis base-catalyzed cycloaddition of electron-deficient activated allenes has been broadly investigated.However, due to the high activation barrier, the Lewis base-catalyzed cycloaddition reaction of nonactivated or weakly activated allenes has rarely been reported.In 2020, Xia and Yu developed the DMAP-catalyzed amino-acylation of arylallenes, providing 2-methyl-3-aroylindole products in good yields [74].Compared with traditional amine moieties as waste after C(O)-N bond cleavage, both acyl and amine moieties are incorporated into the productsviaselective cleavage of amide C-N bonds in the reaction.The experimental results suggested that the Boc group was essential for successful C-N bond cleavage, and no C-N bond cleavage was detected in the absence of N-Boc protection.Further mechanistic studies showed that the amino acylation of allenes occurred in an intramolecular fashion,successive [1,4]- and [1,6]-proton transfers occurred in the recycling system, and a nitrogen anion intermediate assisted the proton transfer processesviaprotonation/deprotonation (Scheme 33).
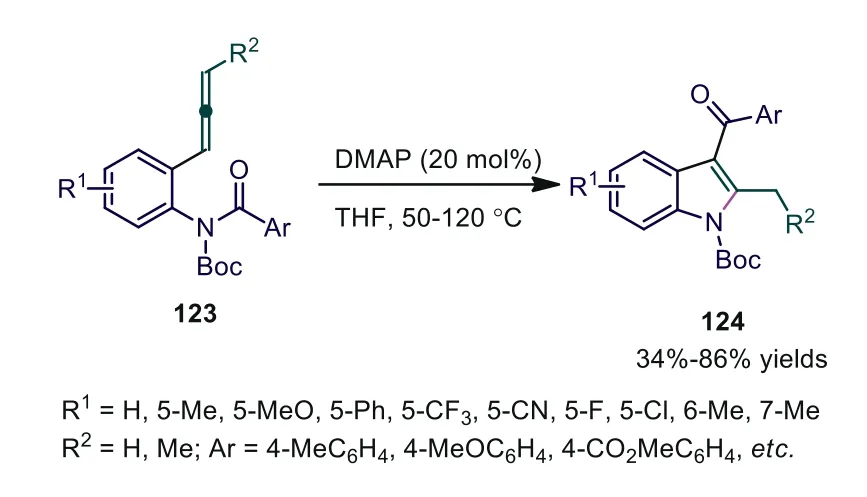
Scheme 33.DMAP-catalyzed amino-acylation of arylallenes.
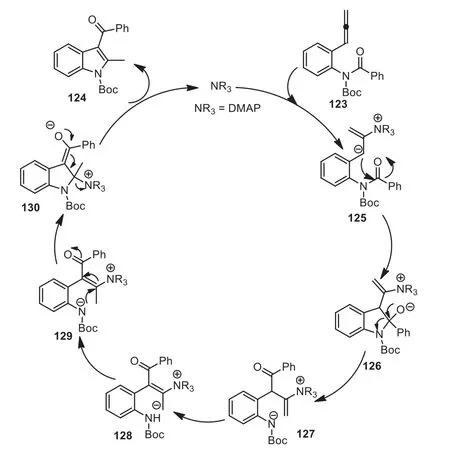
Scheme 34.A plausible reaction pathway of DMAP-catalyzed amino-acylation of arylallenes.
According to control experiments and DFT calculations, the authors proposed a plausible reaction pathway.The nucleophilic addition of DMAP to aryl allene 123 first occurred, affording zwitterionic intermediate 125, which then underwent nucleophilic addition to the amide’s carbonyl to give intermediate 126.Subsequently, successive C-N bond cleavage (127)/[1,4]-(128)/[1,6]-proton transfer (129)/nucleophilic addition of a nitrogen anion to theβposition of theα,β-unsaturated ketone resulted in intermediate 130.Finally, expulsion of DMAP produced product 124 (Scheme 34).
6.Conclusions
Tertiary amine catalysis has emerged as a prominent and reliable strategy for constructing functionalized mono-, multi- and spiro-cyclic structures that have important applications in the fields of natural products synthesis and pharmaceutical design.In the presence of tertiary amines, allenoates can be applied as C2,C3 and C4 synthons to participate in many kinds of cycloaddition reactions, such as [4+2], [2+2], [3+2], [3+3], or sequential annulation reactions.In this review, we have summarized the latest progress of tertiary amine-catalyzed annulation reactions involving diverse allenoates.We hope that this updated review provides our readers with a systematic overview of this research field so that recent advances in tertiary amines catalytic annulation reactions can be better appreciated and recognized.More importantly, we hope that tertiary amine catalysis will continue to evolve and provide even more powerful and versatile synthetic methods to advance the state-of-the-art of ring construction strategies, broadening the horizons of organocatalysis.Thus it is reasonable to believe that new advances in tertiary amine-promoted cycloaddition involving allenoates will come.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper
Acknowledgments
We are grateful to the National Natural Science Foundation of China (No.21702189), Key Scientific and Technological Project of Henan Province (No.202102310004), and Zhengzhou University(No.JC21253007) of China for financial support.
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction