
Advances in cycloaddition and hydroaddition reaction of α-(trifluoromethyl)styrenes without defluorination:An alternative approach to CF3-containing compounds
2022-06-20 07:59YupinDengJingjingHeSongCoXuhongQin
Chinese Chemical Letters 2022年5期
Yupin Deng, Jingjing He, Song Co,*, Xuhong Qin,b,*
a Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology (ECUST), Shanghai 200237, China
b School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
Keywords:α-(Trifluoromethyl)styrenes Cycloaddition and hydroaddition Nondefluorination CF3-containing compounds
ABSTRACT α-(Trifluoromethyl)styrene and its derivatives have found wide applications in the fields of pharmaceuticals, agrochemicals, and advanced materials.They are also versatile trifluoromethyl-containing building blocks for the preparation of various trifluoromethyl-containing, fluorine-containing or nonfluorinated compounds.Recently, great efforts have been made to develop diverse reactions for rapidly accessing a wide range of valuable gem–difluoroalkenes and gem–difluoroalkylated compounds via defluorinative reaction or the defluorinative ipso-functionalization reaction of α-(trifluoromethyl)styrenes, respectively.In contrast,α-(trifluoromethyl)styrenes remain notably underdeveloped with respect to their use in cycloaddition and hydroaddition reaction with retaining of three C–F bonds.This short review herein is aimed to summarize the recent progress on the cycloaddition and hydroaddition reaction including nucleophilic,radical and transition metal-catalyzed addition of α-(trifluoromethyl)styrenes without accompanying defluorination.
1.Introduction
α-(Trifluoromethyl)styrenes have received a great deal of attention due to their important applications in the fields of pharmaceuticals, agrochemicals and functional materials [1,2].They are also versatile and useful trifluoromethyl-containing building blocks for the preparation of a diverse range of trifluoromethylbearing, fluorine-bearing and nonfluorinated organic compounds[3–7].As a consequence, remarkable progress has been achieved in the transformation ofα-(trifluoromethyl)styrenes to various structurally novel molecules [8,9].However, especially in recent years,most of the research efforts have been devoted towards the conversion ofα-(trifluoromethyl)styrenes intogem–difluoroalkenes andgem–difluoroalkylated compoundsviacleavage of the C–F bonds in the trifluoromethyl group (Schemes 1a–e) [10–14].On the contrary,the incorporation of the CF3group inα-(trifluoromethyl)styrenes into the final products withoutβ-fluoride elimination remains largely unexplored.It is probably due to the fact that the C–F bond inα-(trifluoromethyl)styrenes is more easily cleaved in the presence of nucleophiles, nucleophilic radicals, transition-metal nucleophiles and base with formation ofgem–difluoroalkenes and metal fluorides.Although there are several excellent reviews focusing on the defluorination ofα-(trifluoromethyl)styrenes to generategem–difluoroalkenes [15–18], no review articles have been published on the cycloaddition and hydroaddition ofα-(trifluoromethyl)styrenes.On the other hand, the nucleophilic addition products,α-CF3arylethyl group-containing compounds,have also found wide use in pharmaceutical and agrochemical sciences (Fig.1) [19–23].With the above considerations in mind,in this short review, we highlight the most recent advances in the cycloaddition and nucleophilic, radical and transition metalcatalyzed addition ofα-(trifluoromethyl)styrenes without accompanying defluorination (Schemes 1f–i).It should be noted that this review concentrated on the cycloaddition and hydroaddition ofα-(trifluoromethyl)styrenes, other aliphatic 2-trifluoromethyl-1-alkenes and trifluoromethyl-polyfluoroalkenes derivatives will not be included in this review.In addition, this review will focus on the most recent developments of the cycloaddition and hydroaddition ofα-(trifluoromethyl)styrenes during the past two decades but mention to previous work when it is needed.
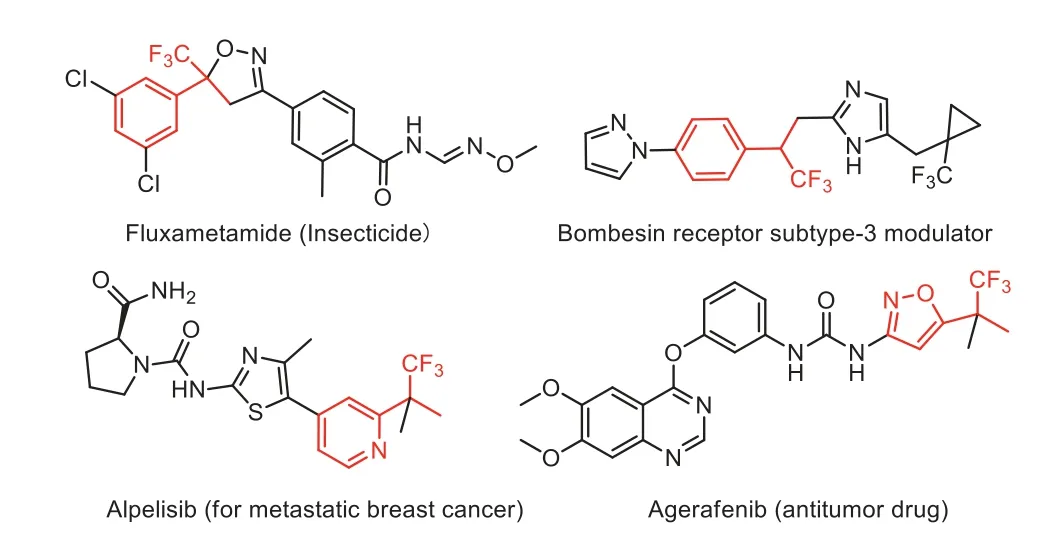
Fig.1.Representative examples of bioactive α-CF3 arylethyl group-containing compounds.
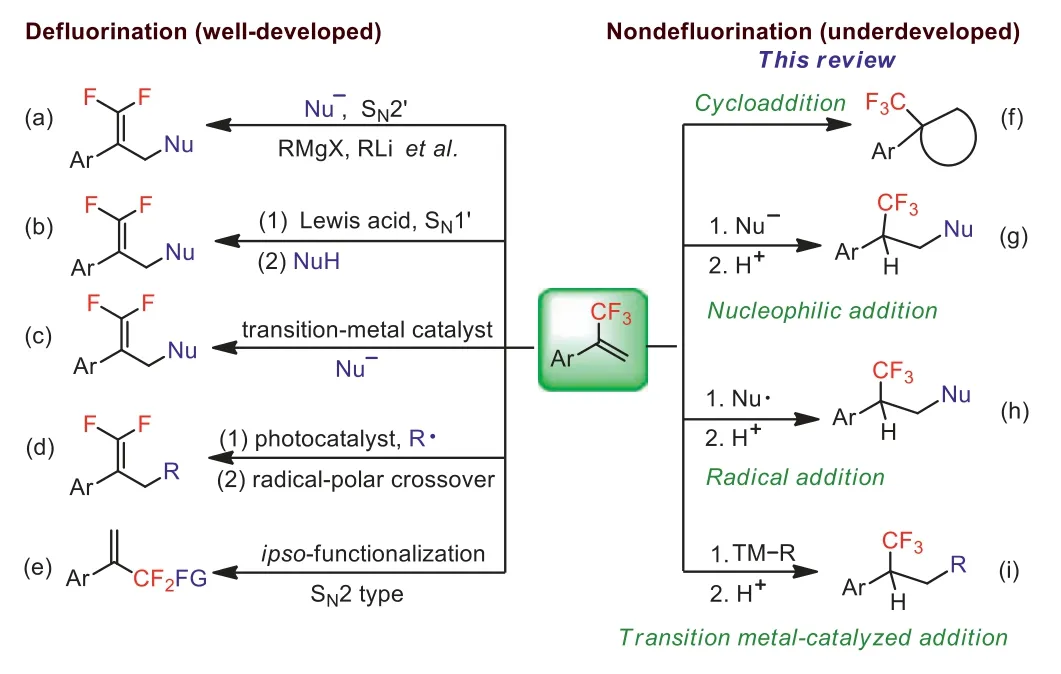
Scheme 1.Typical defluorination and fluorine-retentive reaction.
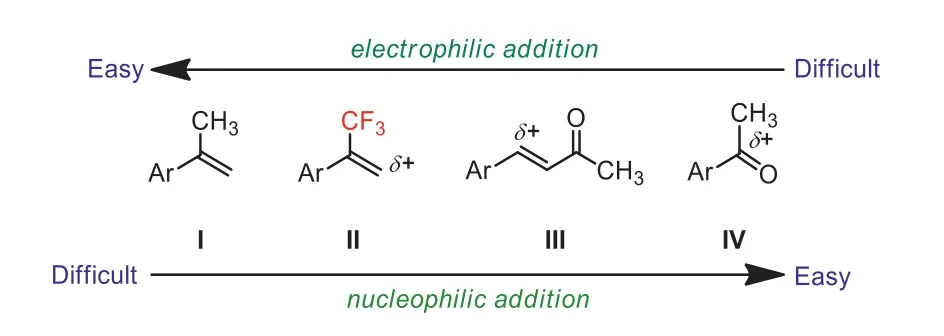
Scheme 2.The comparison electrophilic and nucleophilic reactivities of compounds I–IV.
2.A brief discussion on the electronic properties of α-(trifluoromethyl)styrenes
Generally, alkenes (such as I,α-methylstyrene, Scheme 2) are considered as nucleophiles and therefore they could be easily reacted with electrophiles.α-(Trifluoromethyl)styrene (II, Scheme 2)is activated alkene owing to theσ-electron-withdrawing effect of trifluoromethyl group.The electronic characteristics of the trifluoromethyl group have significant implications on the vinylic terminal carbon ofα-(trifluoromethyl)styrene.Consequently, the terminal carbon atom ofα-(trifluoromethyl)styrene is much more electron-poor than the correspondingα-methylstyrene arene I.The trifluoromethyl group on the carbon-carbon double bond can significantly decrease the LUMO energy level of the alkene [24,25].On the other hand, compared to other relatively strong electrophiles such asα,β-unsaturated ketone and ketone (III and IV, Scheme 2),α-(trifluoromethyl)styrene is slightly less reactive toward nucleophilic attack due to the absence ofπ-electron-withdrawing group(for example, ketone, ester, cyano, or nitro).These observations might be ascribed to the fact that theπ-LUMO energies are lowered by attachment to a carbonyl group more than by attachment to CF3group.Based on the above-mentioned considerations, we envision thatα-(trifluoromethyl)styrenes could be used as the moderate electrophiles and a nucleophilic attack is expected to occur at the methylene terminus of the olefin.Althoughα-(trifluoromethyl)styrenes are appropriate electrophiles with enough reactivity toward nucleophiles, most resulting products aregem–difluoroalkenes andgem–difluoroalkylated compounds which are formed byviacleavage of the C–F bonds in the trifluoromethyl group.It is because theα-(trifluoromethyl)styrenes are susceptible toβ-fluoride elimination when they react with nucleophiles in the presence of base or metal catalyst.In contrast, hydroaddition ofα-(trifluoromethyl)styrenes with nucleophiles without accompanying defluorination has scarcely been explored.
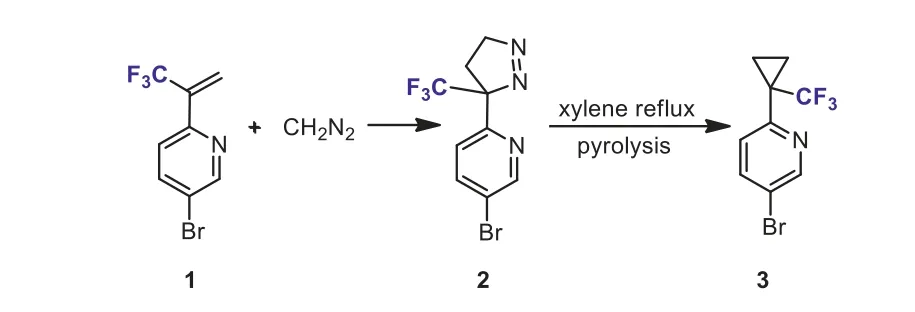
Scheme 3.Cycloaddition of 2-(1-trifluoromethylvinyl)pyridine 1 with diazomethane.
Scheme 4.Fe-catalyzed cycloaddition of α-(trifluoromethyl)styrene with ethyl 2-diazoacetate.
3.Cycloaddition of α-(trifluoromethyl)styrene without breaking a C–F bond
Cycloaddition ofα-(trifluoromethyl)styrene with different substrates is an efficient and practical strategy for the synthesis of novel trifluoromethyl-containing heterocycles and carbocycles.Although the defluorination products are seldom observed during the cycloaddition process, the use ofα-(trifluoromethyl)styrene in cycloaddition reaction for the preparation of the trifluoromethylcontaining cyclic compounds is still underdeveloped.In this section, we extensively reviewed the cycloaddition ofα-(trifluoromethyl)styrenes with various substrates.
3.1.Synthesis of trifluoromethyl-substituted three-membered rings
Trifluoromethyl-substituted cyclopropane (TFCp) is an important structural motif found in bioactive compounds.It is reported that CF3-substituted cyclopropane could be considered as a bioisostere oftert–butyl group and could increase metabolic stability of trifluoromethylated cyclopropyl group-containing molecules[26].Up to now, numerous methods for the synthesis of CF3-substituted cyclopropanes have been developed [27].One of the most common methods for the synthesis of CF3-substituted cyclopropanes involves the [3 + 2] cycloaddition of trifluoromethylsubstituted alkenes such as 2-trifluoromethylacrylates with diazoalkanes followed by a thermally-induced ring contraction[28].However, the use ofα-(trifluoromethyl)styrene as reactant has rarely been reported in the literature.For example, Barnes-Seeman reported the [3 + 2] cycloaddition of 2-(1-trifluoromethylvinyl)pyridine 1 with diazomethane [29].The pyrolysis of the resulting 1-pyrazoline 2 at 140 °C afforded trifluoromethylcyclopropyl-containing pyridine 3 (Scheme 3).
Scheme 5.Fe-catalyzed cycloaddition of α-(trifluoromethyl)styrene with diazo acetonitrile.
Scheme 6.Rh-catalyzed asymmetric cyclopropanation of α-trifluoromethyl styrenes with α-aryl diazo acetates and ethyl α-cyanodiazo acetate.
Scheme 7.Reaction of α-(trifluoromethyl)styrene with three halomethyl-based silicate reagents.
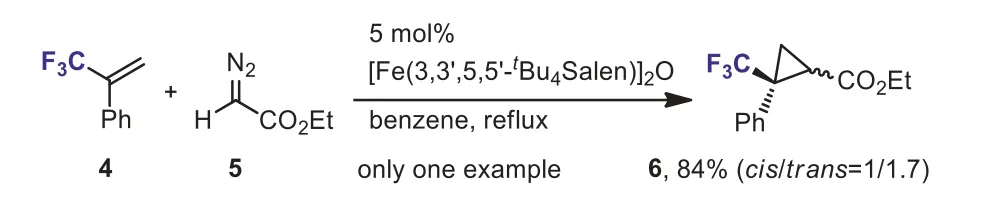
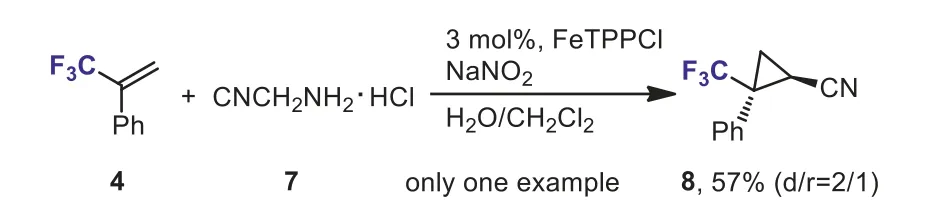
In 2003, Nguyenet al.reported Fe-catalyzed cycloaddition ofα-(trifluoromethyl)styrene 4 with ethyl 2-diazoacetate 5.A mixture ofcis- andtrans-isomers of cyclopropanes 6 was obtained in 84% yield (Scheme 4) [30].However, only one example was described.The authors also indicated that the yield decreased dramatically and only 28% of 6 was obtained in absence of iron(III) complexes.One example of Fe-catalyzed cycloaddition ofα-(trifluoromethyl)styrene 4 with diazo acetonitrile, generatedin situfrom amine 7, has also been reported (Scheme 5).A trifluoromethyl and nitrile-substituted cyclopropane 8 was formed in moderate yield [31].
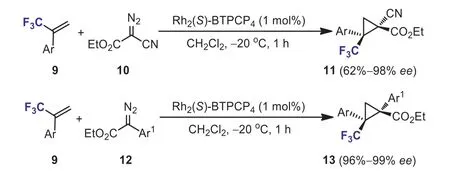
In 2018, Charette and Jubaultet al.developed a general and effi-cient access to chiral functionalized trifluoromethyl cyclopropanes 11 and 13viathe enantioselective Rh-catalyzed cyclopropanation ofα-trifluoromethyl styrenes 9 with ethylα-cyanodiazo acetate 10 andα-aryl diazo acetates 12, respectively (Scheme 6) [32].The cyclopropanation proceeded smoothly at -20 °C in the presence of 1 mol% Rh2(S)-BTPCP4, affording various polyfunctionalized trifluoromethylated cyclopropanes in moderate to high yields with high diasteroselectivities and enantioselectivities.In addition, the synthetic utility of the valuable trifluoromethylated cyclopropanes was demonstrated by derivatization reactions and several useful enantiomerically pure cyclopropanes were synthesized efficiently.
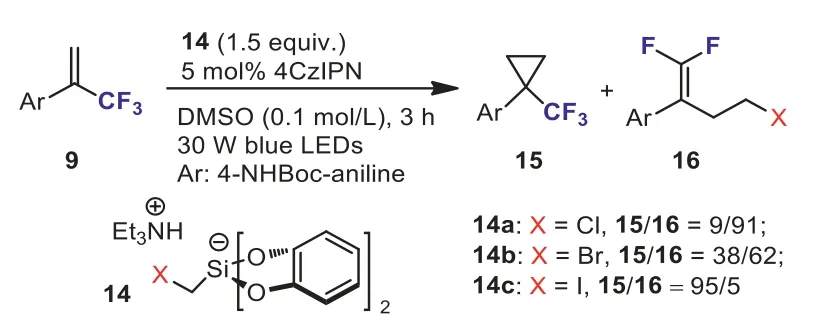
Single-electron access to cyclopropanes still remains underdeveloped.No photooxidizable C1 reagent for radical cyclopropanation has emerged in the literature.To bridge this gap, in 2018, Gutierrez and Molander’s group designed and synthesized three novel halomethyl-based silicate reagents (14a–14c)(Scheme 7) [33].During the course of screening the reaction conditions for the redox-neutral cyclopropanation of olefins, they found that the distribution of trifluoromethyl-substituted cyclopropane 15 and undesiredgem–difluoroalkene 16 was highly dependent on the halomethyl silicate employed and iodomethylbased silicate reagent 14c could provide the expected cyclopropanation product 15 in excellent yield.Subsequently, the scope and limitations of the novel redox-neutral cyclopropanation ofα-(trifluoromethyl)styrenes 9 with iodomethylsilicate 14c were investigated (Scheme 8).This reaction exhibited a broad substrate scope and a variety of functional groups were well tolerated under the optimized reaction conditions, providing trifluoromethylsubstituted cyclopropanes 15 in moderate to good yields.Based on the results of theoretical and experimental mechanistic investigation, they indicated that the reaction likely proceedsviaan anionic 3-exo-tetring closure.
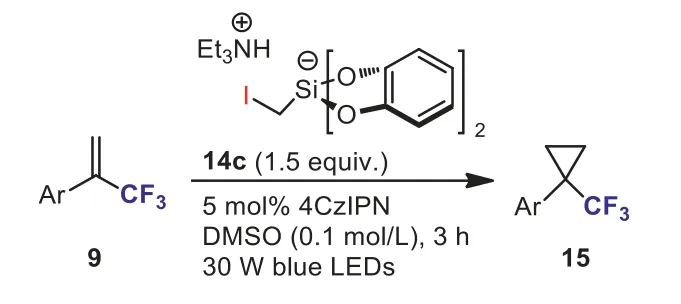
Scheme 8.Cyclopropanation of α-(trifluoromethyl)styrenes with iodomethylsilicate.
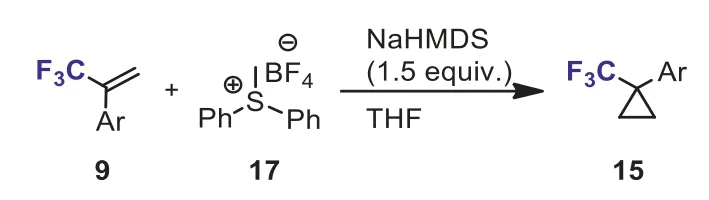
Scheme 9.Cyclopropanation of 1-aryl-1-trifluoromethyl ethylenes with sulfonium ylide.
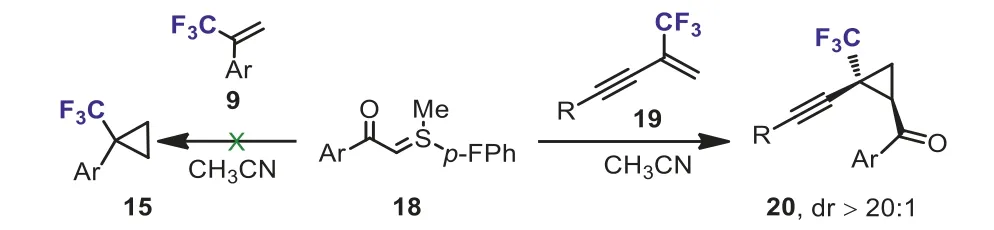
Scheme 10.Cyclopropanation of trifluoromethylenynes and α-(trifluoromethyl)styrene with aroyl sulfur ylides.
The Johnson–Corey–Chaykovsky reaction is an alternative and convenient approach to cyclopropane.For example, Xiaoet al.used(2,2,2-trifluoroethyl)diphenylsulfonium triflate as an efficient ylide reagent for the Johnson–Corey–Chaykovsky reaction [34].The cyclopropanation ofα,β-unsaturated ketones with sulfonium salt(Ph2S+CH2CF3OTf-) could affordtrans-trifluoromethyl and carbonyl group-containing cyclopropanes in high yields.Inspired by the Johnson–Corey–Chaykovsky reaction involving sulfonium ylide, recently, Cyr and Marinieret al.described a mild and diazo-free method for the synthesis of trifluoromethyl cyclopropanes by the cyclization of 1-aryl-1-trifluoromethyl ethylenes 9 with sulfonium ylide (Ph2SMeBF4, 17).In most cases, trifluoromethyl alkenes bearing electron-neutral, -rich and -poor rings could provide the corresponding CF3-cyclopropanes 15 in good yields.This strategy may provide an alternative approach to prepare useful trifluoromethylcyclopropanes (Scheme 9) [35].
In addition, another specific example of cyclopropanation reaction needs to be mentioned is that the 2-trifluoromethyl-1,3-enynes are capable of undergoing ylide cyclopropanation.In 2019, Liu and Wanget al.reported a highly diastereoselective cyclopropanations of trifluoromethylenynes 19 with aroyl sulfur ylides 18.A variety of CF3-substituted cyclopropanes containing alkynyl and carbonyl groups 20 were synthesized under mild reaction conditions (Scheme 10) [36].However, whenα-(trifluoromethyl)styrene 9 was used as substrate, no cyclopropanation product 15 was obtained.The authors speculated that the electron-withdrawing character of the alkynyl group might further lower the LUMO energy level of trifluoromethylenyne.They also indicated that no fluoride elimination was observed.
More recently, Koenigset al.developed a novel protocol for the photocatalytic synthesis of trifluoromethylated aziridines 22 by Ru-catalyzed direct aziridination reactions ofα-trifluoromethyl styrene with iminoiodinane (21, PhINTs) under visible-light irradiation from 3 W blue LEDs (470 nm) (Scheme 11) [37].The reaction exhibited good substrate scope and functional group compatibility.The authors indicated that the nitrene radical anion 23 served as a reactive intermediate in the aziridination reaction.
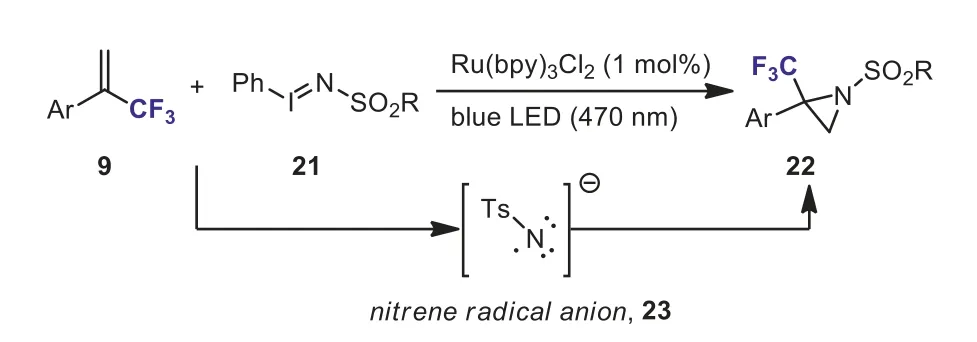
Scheme 11.Ru-catalyzed aziridination reactions of α-trifluoromethyl styrene with iminoiodinane.
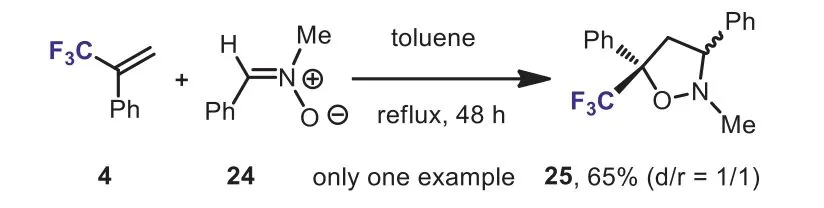
Scheme 12.Cycloaddition of nitrone with α-(trifluoromethyl)styrene.
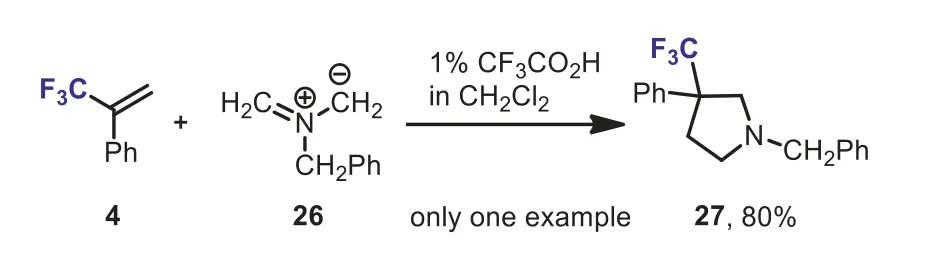
Scheme 13.Cycloaddition of azomethine ylide with α-(trifluoromethyl)styrene.
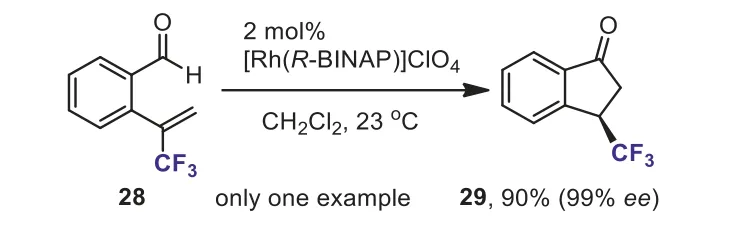
Scheme 14.Asymmetric hydroacylation of 2-formyl α-(trifluoromethyl)styrene.
3.2.Synthesis of trifluoromethyl-substituted five- and six-membered rings
The important application of theseα-(trifluoromethyl)styrenes has been in their use as dipolarophiles for 1,3-dipolar cycloaddition.Althoughα-(trifluoromethyl)styrenes are electron-deficient alkenes, generally, they are not good dipolarophiles for 1,3-dipolar cycloaddition due to the absence of a strong electronwithdrawingπ-acceptor such as cyano, ester, ketone, nitro or sulfone.Therefore, the use ofα-(trifluoromethyl)styrene as dipolarophile to undergo the 1,3-dipolar cycloaddition is rare.In consideration of the strong electron-withdrawing nature of the CF3group which could decrease the LUMO energy levels of the alkenes,α-(trifluoromethyl)styrenes were expected to act as dipolarophile in [3 + 2] cycloaddition reactions [24,25].
As early as 1991, Béguéet al.investigated the 1,3-dipolar cycloaddition between nitrone (N-(benzylidene)-methylamine-Noxide 24 andα-(trifluoromethyl)styrene 4.A diastereoisomeric mixture of trifluoromethyl isoxazolidines 25 was obtained in 65%yield (Scheme 12) [38].
Two years later, Béguéet al.studied the 1,3-dipolar cycloaddition of non-stabilized azomethine ylide 26 with several trifluoromthylated olefins.They foundα-(trifluoromethyl)styrene 4 was also a good dipolarophile and could afforded polysubstituted 3-trifluoromethyl-pyrrolidine 27 in 80% yield (Scheme 13) [39].
Moreheadet al.developed Rh-catalyzed asymmetric hydroacylation of 2-formylα-(trifluoromethyl)styrene 28 and a chiral 3-trifluoromethyl indanone 29 was synthesized in high yield with 99%ee(Scheme 14) [40].Unfortunately, only one example was reported.
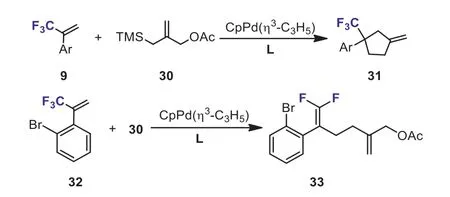
Scheme 15.Pd-catalyzed [3 + 2] cycloaddition or defluorination of α-(trifluoromethyl)styrenes with trimethylenemethane.
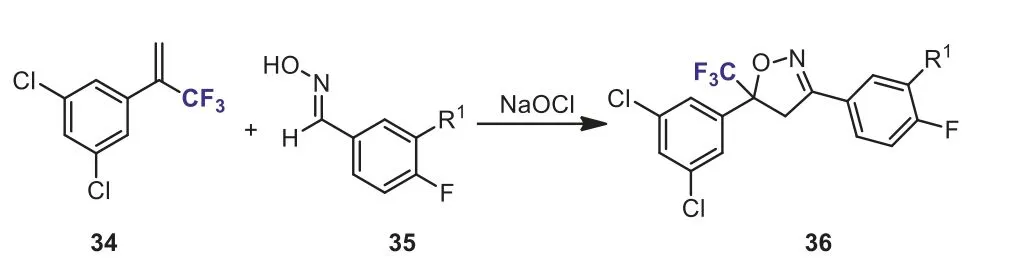
Scheme 16.Cycloaddtion of α-(trifluoromethyl)styrene with oxime.
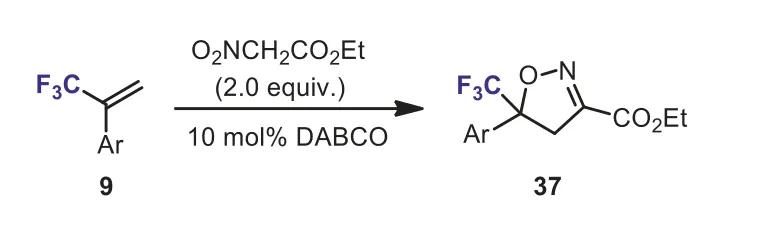
Scheme 17.Cycloaddtion of α-(trifluoromethyl)styrene with ethyl nitroacetate.
In 2015, the group of Trost has demonstrated the first example of the palladium-catalyzed [3 + 2] cycloaddition of trimethylenemethane (30, TMM) withα-(trifluoromethyl)styrenes.This novel method allows for the preparation of various exomethylene cyclopentanes bearing a quaternary center substituted by the trifluoromethyl group 31.Theσ-electron-withdrawing trifluoromethyl group played a key role in this cycloaddition.In addition, when 2–bromoα-(trifluoromethyl)styrene 32 was used as substrate, undesiredβ-fluorine elimination occurred andgem–difluoroalkene 33 was formed exclusively (Scheme 15) [41].
Recently, much attention has been paid to the synthesis of trifluoromethyl-containing isoxazoline derivatives due to their wide applications in pesticide chemistry.Treatment ofα-(trifluoromethyl)styrene 34 with oxime 35 in the presence of NaOCl would produce isooxazoline 36 (Scheme 16), which is the key intermediate of insecticide [42].Another method for the synthesis of CF3-containing isoxazolines was the cyclization ofα-(trifluoromethyl)styrene 9 with ethyl nitroacetate in the presence of a catalytic amount of DABCO (1,4-diazabicyclo[2.2.2]octane).The reaction was carried out in anhydrous EtOH at 80 °C for 80 h and the target products, ethyl 5-aryl-5-(trifluoromethyl)-1,2-dihydroisoxazole-3-carboxylates 37 were obtained in 65%–75%yields (Scheme 17) [43].
In 2017, Pozo and Fusteroet al.described an intramolecular 1,3-dipolar cycloaddition ofortho-substitutedα-(trifluoromethyl)styrene-derived nitrones 39, which were generatedin situfromα-(trifluoromethyl)styrenes bearing an aldehyde functionality in theorthoposition 38 andN-alkylhydroxylamine hydrochloride.Tricyclic fused isoxazolidines bearing a quaternary trifluoromethyl moiety 40 were obtained as major or exclusive products, whereas the bridged cycloadducts were formed as by-products 41 (Scheme 18) [44].They also suggested that the role of the trifluoromethyl group is crucial in the regioselectivity of the reaction, leading to the formation of fused isoxazolidines preferentially or exclusively.
In continuation of their interest in new cycloaddition ofα-(trifluoromethyl)styrenes bearing an aldehyde in theorthoposition 38, Pozo and Fusteroet al.disclosed a novel method for the synthesis of tri- and tetracyclic fused pyrrolidines bearing a quaternary trifluoromethyl group 43 by means of an intramolecular azomethine ylide 44 cycloaddition with trifluoromethylated dipolarophiles 38.The intermediates 44, azomethine ylides (AMY),were generatedin situfrom the reaction of 38 withα-amino acids 42 in toluene.Intramolecular cycloaddition of azomethine ylides could afford tricyclic and tetracyclic pyrrolidines in moderated yields in a stereoselective manner (Scheme 19) [45].

Scheme 18.Cycloaddition of α-(trifluoromethyl)styrenes bearing an aldehyde functionality in the ortho position.
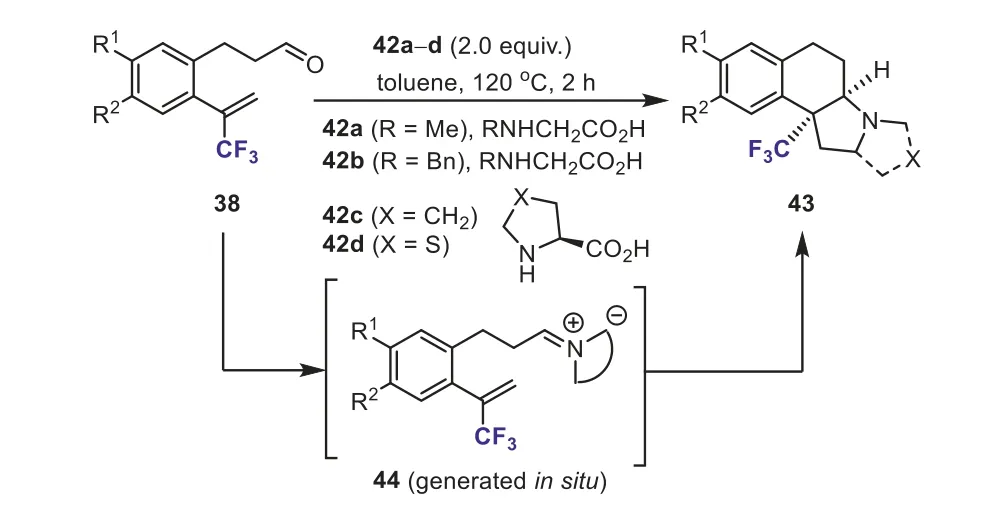
Scheme 19.Synthesis of tricyclic and tetracyclic pyrrolidines from trifluoromethylcontaining aldehydes and α-amino acids.

Scheme 20.The 1,3-dipolar cycloaddition of α-(trifluoromethyl)styrene with nitroalkane.
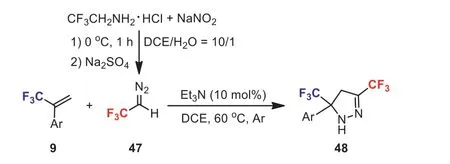
Scheme 21.[3 + 2] Cycloaddition reactions of 2,2,2-trifluorodiazoethane with α-(trifluoromethyl)styrenes.
In 2020, Tsubakiet al.usedα-(trifluoromethyl)styrene 4 as dipolarophile to react withO-alkyloxime-substituted nitroalkanes 45.The 1,3-dipolar cycloaddition reaction proceeded efficiently in the presence of dehydrating reagent PhNCO, affording trifluorinated 2-isoxazoline 46 in high yield (Scheme 20) [46].
More recently, our group developed an efficient method for the synthesis of bis(trifluoromethyl)-substituted pyrazolines 48via[3 + 2] cycloaddition reactions of 2,2,2-trifluorodiazoethane 47 withα-(trifluoromethyl)styrenes 9 in the presence of a catalytic amount of Et3N (Scheme 21) [47].A range of bis(trifluoromethyl)-substituted 2-pyrazolines were obtained in good to excellent yields.This method also exhibits wide functional group compatibility.The results indicated thatα-(trifluoromethyl)styrenes could be used as excellent dipolarophile partners in [3 + 2] dipolar cycloaddition [48,49].
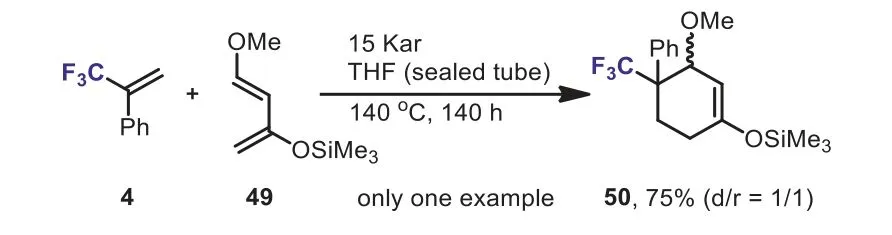
Scheme 22.[3 + 2] Cycloaddition reactions of 2,2,2-trifluorodiazoethane with α-(trifluoromethyl)styrenes.
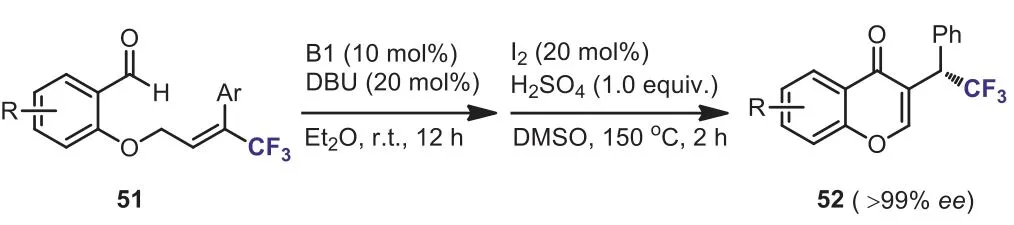
Scheme 23.Cyclization of CF3 substituted o-allyloxybenzaldehyde.
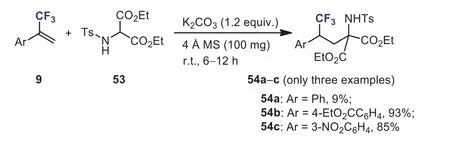
Scheme 24.Nucleophilic addition of α-(trifluoromethyl)styrenes with N-tosylated 2-aminomalonates.
α-(Trifluoromethyl)styrenes could also be used as substrates for the synthesis of six-membered rings.For example, in 1992, Béguéet al.reported the preparation of CF3-substituted cyclohexene 50 by the Diels-Alder reaction of Danishefsky’s diene 49 withα-(trifluoromethyl)styrene under high pressure (Scheme 22) [50].For comparison purpose, whenα-methylstyrene was used as substrate,no cycloaddition occurred and polymeric material was formed.
In 2018, Chenget al.disclosed a useful method for the synthesis of enantiopure trifluoromethyl-substituted chromenonesviaa one-pot carbene-catalyzed umpolung/oxidation process of CF3substitutedo-allyloxybenzaldehyde 51.A variety of enantiopure chromenones having trifluoromethylated stereogenic center in theβposition of the carbonyl group 52 was synthesized in moderate to good yields with excellent enantioselectivities (Scheme 23) [51].
4.Hydroaddition of α-(trifluoromethyl)styrenes without accompanying defluorination
Recently, Altman and co-workers have extensively investigated the nucleophilic hydrofunctionalization reactions ofgem–difluoroalkenes without affecting the C–F bond.This fluorineretentive strategy provides a facile and efficient alternative for the preparation of the difluorinated compounds [52,53].While hydroaddition ofα-(trifluoromethyl)styrenes withoutβ-fluoride elimination is still in its infancy in terms of reaction efficiency,yield and types.Although a few examples involving the hydroaddition of (trifluoromethyl)styrenes have been reported, but examples that delivers the hydroaddition products as the main products are rare.In most cases, hydroaddition products are observed or formed as by-products.In some other cases, the formation of the defluorination products and hydroaddition products depends significantly on the reaction conditions used.
4.1.The hydroaddition products as the main products
In 2016, Zhanget al.found that in contrast to the results of the reaction ofα-trifluoromethyl-α,β-unsaturated carbonyl compounds with nucleophiles,N-tosylated 2-aminomalonates 53, no defluorination products were observed whenα-(trifluoromethyl)styrenes were used, instead, the unexpected nucleophilic addition products 54 were obtained as the sole product (Scheme 24) [54].α-(Trifluoromethyl)styrenes bearing strong electron-withdrawing 54b and 54c had a beneficial effect on the yield of the addition reaction.When (3,3,3-trifluoroprop-1-en-2-yl)benzene was used as substrate, the yield of addition product 54a decreased to 9%.
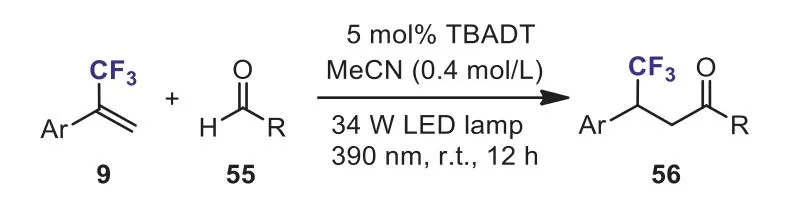
Scheme 25.The photocatalytic hydroacylation of α-(trifluoromethyl)styrenes via nucleophilic acyl radical.
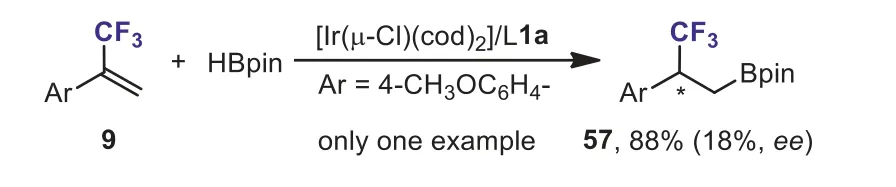
Scheme 26.The Ir-catalyzed hydroboration of α-(trifluoromethyl)styrene with HBpin.
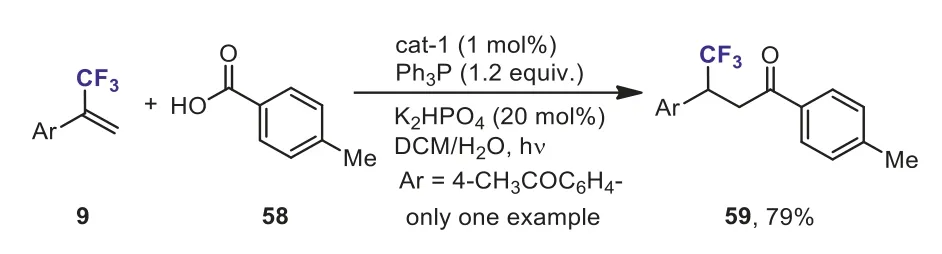
Scheme 27.The Ir-catalyzed deoxygenative C–C coupling of aromatic carboxylic acid with α-(trifluoromethyl)styrene.
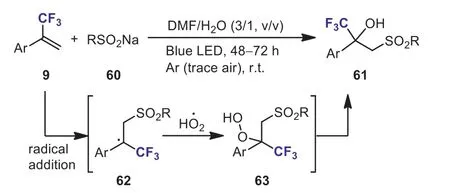
Scheme 28.The visible-light-driven sulfonation of α-(trifluoromethyl)styrenes.
In 2019, Wanget al.developed a versatile and powerful approach to valuableβ-CF3ketones 56 through the photocatalytic hydroacylation ofα-(trifluoromethyl)styrenes with aldehydes 55 (Scheme 25) [55].The tetrabutylammonium decatungstate(TBADT) was used as a hydrogen-atom-transfer (HAT) photocatalyst for acyl C–H activation.The TBADT-catalyzed hydroacylation method offered several remarkable advantages including high functional group tolerance, excellent regioselectivity and complete atom economy.Under the optimized condition, theβ-elimination was effectively suppressed.
One example of iridium-catalyzed asymmetric hydroboration ofα-(trifluoromethyl)styrene 9 with HBpin has been reported by Pàmies and Diéguez (Scheme 26) [56].In contrast to other 1,1-disubstituted aryl olefins, which provided the hydroboration products with high enantioselectivity,α-(trifluoromethyl)styrene 9 gave the hydroborated product 57 with poor enantioselectivity.
During the course of their investigation on the direct deoxygenative ketone synthesis from aromatic carboxylic acids 58 and alkenes in the presence of photocatalyst, Zhu’s group found thatα-(trifluoromethyl)styrene was also a good coupling partner and afforded the trifluoromethylated ketone 59 in good yield(Scheme 27) [57].
In 2021, Xiang and Yanget al.reported a visible-light-induced sulfonation ofα-(trifluoromethyl)styrenes 9 with sodium sulfinates 60.A variety of structurally diverse novelα-trifluoromethylβ-sulfonyl tertiary alcohols 61 were obtained in good yields(Scheme 28) [58].The authors proposed a CTC (charge-transfer complexes)-involved mechanism for this photoinduced process.The radical RSO2·was added to the double bond ofαtrifluoromethylstyrenes to give the trifluoroalkyl radical species 62.Radical cross-coupling of radical 62 and superoxide anion radical(HO2·) led to peroxide intermediate 63.The cleavage of the peroxide bond of 63 furnished the desired products 61.
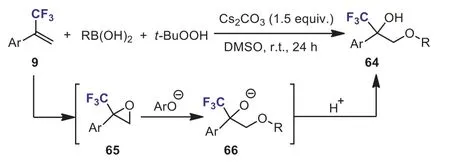
Scheme 29.TBHP-enabled regioselective hydroetherification of α-(trifluoromethyl)styrenes with boronic acids.
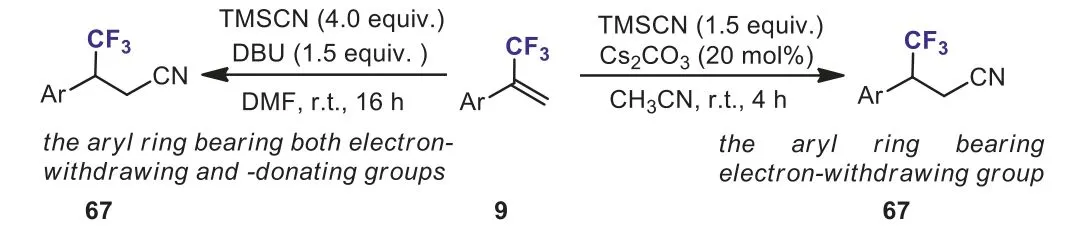
Scheme 30.Synthesis of CF3-containing linear nitriles from α-(trifluoromethyl)styrenes.
TBHP (tert–butyl hydroperoxide)-promoted regioselective 1,2-difunctionalization of alkenes has attracted considerable attention from synthetic chemists.It is an efficient tool for the synthesis of functionalized alcohols.In 2021, Zhuet al.developed a novel and facile method for the synthesis of various usefulα-trifluoromethylβ-aryloxy tertiary alcohols 64 through a regioselective one-pot,three-component reaction ofα-(trifluoromethyl)styrenes, TBHP and boronic acids (Scheme 29) [59].This hydroetherification proceeded smoothly under mild reaction conditions and provided the CF3-containing tertiary alcohols in high yields.TBHP might serve as the oxidant, nucleophile andO-source of the products.The addition oftert–butyl peroxide anion toα-(trifluoromethyl)styrene resulted in the formation of the epoxide intermediate 65.The regioselective nucleophilic ring-opening reaction of epoxide 65 with aryloxy anion (ArO–), which is generatedin situfrom TBHP and boronic acids, would afford the intermediate 66.Further protonation of intermediate 66 gave the expected products 64.
The above-mentioned two bifunctionalization ofα-(trifluoromethyl)styrenes would provide a practical strategy for the preparation of various functionalized CF3-containing compounds.
More recently, our group developed two unprecedented Cs2CO3-catalyzed and DBU-mediated nucleophilic additions of TMSCN toα-(trifluoromethyl)styrenes.The reaction proceeded effi-ciently at room temperature under mild and transition-metal free conditions without accompanying defluorination of trifluoromethyl group.A range of structurally diverse CF3-containing linear nitriles 67 were obtained in moderate to excellent yields in a highly regioselective manner (Scheme 30) [60].
4.2.The generation of hydroaddition and defluorination products depending on the reaction conditions
In 2008, Ichikawaet al.reported the disfavored nucleophilic 5-endo-trigcyclization of 1-(trifluoromethyl)vinyl compounds to yield five-membered hetero- and carbocycles bearing fluorinated one-carbon units.The cyclization ofα-(trifluoromethyl)styrenes bearing a nucleophilic nitrogen atom at theorthoposition 68 in the presence of NaH could afford 3-(difluoromethylene)indolines 69 through intramolecular SN2′ reaction with loss of a fluoride ion.The use of DBU as base resulted in the formation of intramolecular nucleophilic addition products, 3-(trifluoromethyl)indolines 70 (Scheme 31) [61].Furthermore, 3-difluoromethylene 2,3-dihydrobenzo[b]thiophene 72 and 3-trifluoromethyl 2,3-dihydrobenzo[b]thiophene 73 could be obtained by the cyclization of 2-(3,3,3-trifluoroprop-1-en-2-yl)thiophenol ester 71 through the nucleophilic substitution and addition, respectively, depending on the bases and solvents involved.The authors suggested that the sulfonamide NH group of 68 and/or DBU·H+and CH3OH acted as a proton donor.
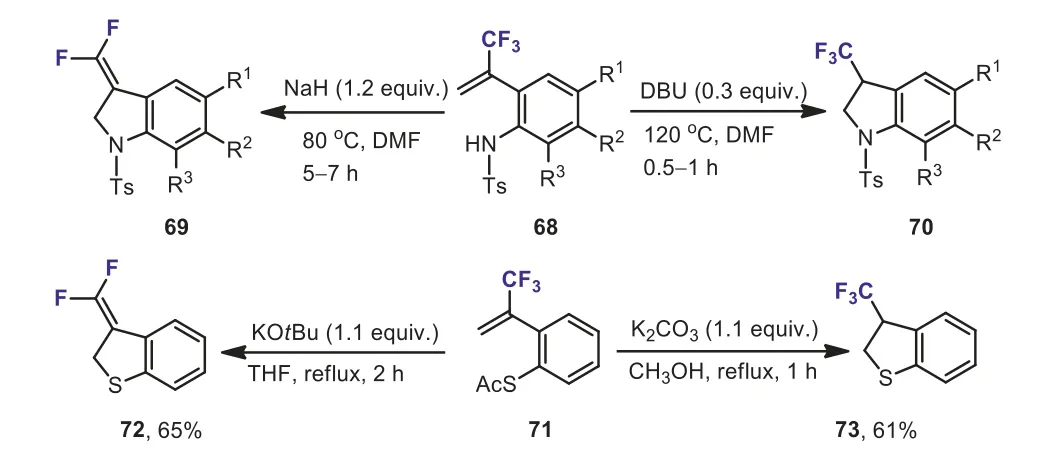
Scheme 31.Nucleophilic 5-endo-trig cyclization of α-(trifluoromethyl)styrenes bearing a nucleophilic nitrogen or sulfur atom at the ortho position.
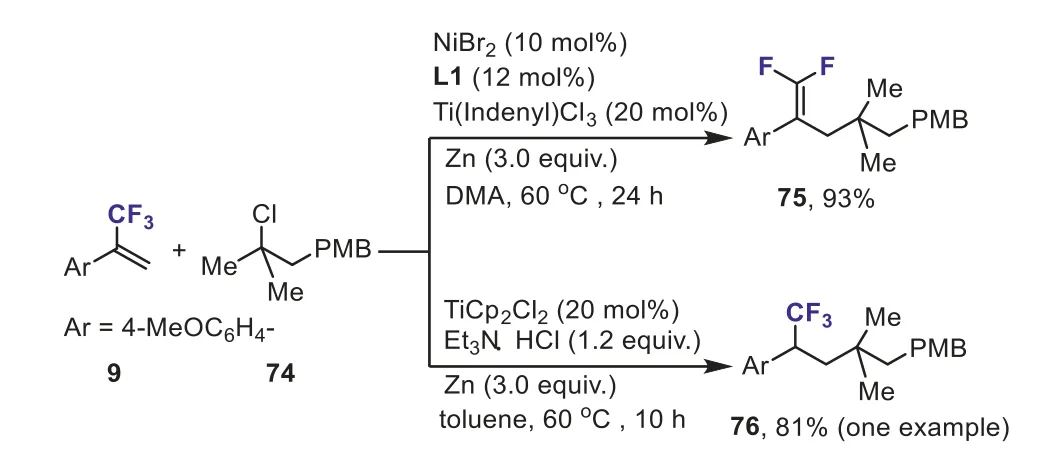
Scheme 32.Reaction of α-(trifluoromethyl)styrene with tertiary alkyl chloride under different reaction conditions.
In 2019, Wanget al.disclosed a Ni, Ti-cocatalyzed reductive allylic defluorinative cross-coupling reaction ofα-(trifluoromethyl)styrenes with unactivated alkyl chlorides and bromides.This new method provided an efficient and highly versatile entry to synthesis of a range ofgem–difluoroalkenes.They also further demonstrated that the reaction ofα-(trifluoromethyl)styrene 9 (Ar = 4-MeOC6H4-) with tertiary alkyl chloride 74 could provide thegem–difluoroalkene 75 (defluorination product) and addition product 76 (nondefluorination product) in good yields,respectively, under different reaction conditions (Scheme 32) [62].
4.3.The formation of hydroaddition products as by-products
As mentioned in the Introduction,α-(trifluoromethyl)styrenes can easily undergoβ-F elimination to afford thegem–difluoroalkenes.Generally,gem–difluoroalkenes are obtained as the sole products, however, in some cases, hydroaddition products are detected as by-products.Considering the low yields of hydroaddition products obtained for the these reported examples,therefore, chances and challenges still remain, such as tuning the reaction conditions and finding proper ways to control the product distribution.
In 2017, Shimakoshi and Hisaedaet al.reported a photocatalytic reduction ofα-(trifluoromethyl)styrenes in the presence of a catalytic amount of B12-TiO2under UV light irradiation.A mixture of the defluorinated product,β,β-difluoro-α-methylstyrenes 77 and the hydrogenated product,α-trifluoromethyl ethylbenzenes 78, was formed (Scheme 33) [63].The DFT calculations revealed that the reaction proceededvia α-radical intermediate 79 and carbanion intermediate 80.
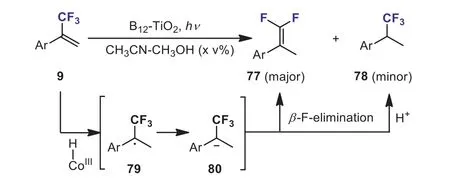
Scheme 33.B12-TiO2-catalyzed reduction of α-(trifluoromethyl)styrenes.
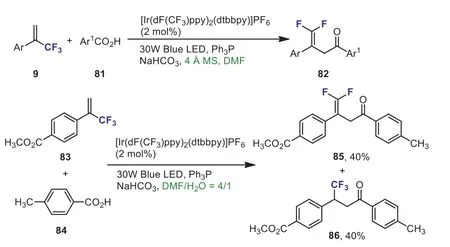
Scheme 34.Reaction of α-(trifluoromethyl)styrenes with carboxylic acids.
In 2020, Wanget al.described a visible-light-induced deoxygenation/defluorination protocol for synthesis ofγ,γdifluoroallylic ketones 82 from aromatic carboxylic acids 81 andα-(trifluoromethyl)styrenes 9.However, the coupling reaction ofα-(trifluoromethyl)styrene 83 with carboxylic acid 84 using water and DMF as solvent, theγ,γ-difluoroallylic ketone 85 and addition product 86 were isolated in 80% total yield as a 1:1 mixture.The presence of water was beneficial for the formation of the trifluoromethyl-containing product (Scheme 34) [64].A plausible mechanism involving the CF3-styrene carbon radical and CF3-styrene carbanion intermediate was suggested.
In 2008, Murakamiet al.reported a rhodium-catalyzed addition reaction of arylboronic esters or phenylboronic acid toα-(trifluoromethyl)styrenes.The alkylrhodium(I) intermediate 87 underwent eitherβ-fluoride elimination to affordgem–difluoroalkene 88 or protodemetalation with phenylboronic acid or H2O to give addition product 89 in the absence of additive (Scheme 35) [65].
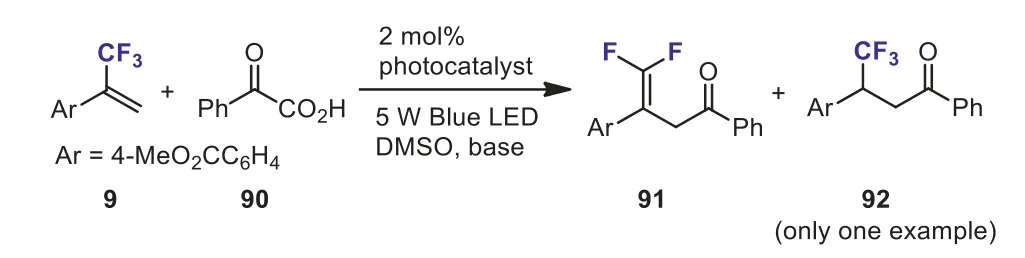
In 2016, Zhouet al.investigated the Ir-catalyzed coupling reaction of 4-ester-substitutedα-(trifluoromethyl)styrene 9 with benzoylformic acid 90 under the irradiation of a 5 W blue LED(Scheme 36) [66].They found that a mixture ofgem–difluoroalkene 91 and nondefluorination product 92 was obtained using 2,6-lutidine as base.Thegem–difluoroalkene 91 was isolated as the sole product by changing 2,6-lutidine to LiOH.The reaction proceeded through a radical-polar crossover mechanism.
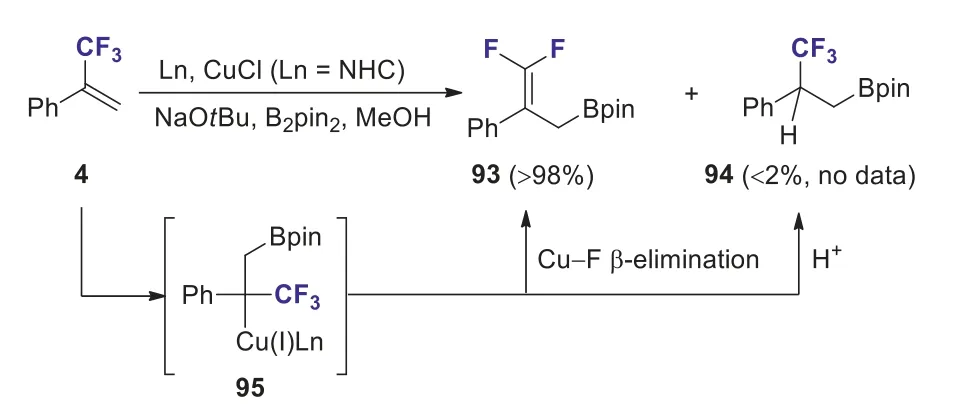
The copper catalyzed defluoroborylation of trifluoromethylalkenes with B2pin2has been well-documented.In 2019, Hoveydaet al.disclosed that the borylation of (3,3,3-trifluoroprop-1-en-2-yl)benzene 4 with B2pin2afforded difluoroallyl boronate 93 in excellent yield (GCMS) should be (GC-MS), along with a small amount of unexpected hydroborylation product 94.The Cu–Fβelimination and protonation of Cu–alkyl intermediate 95 furnished the defluorinative product 93 and CF3-containing product 94, respectively (Scheme 37) [67].
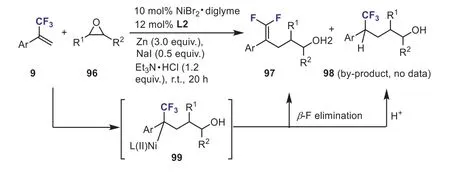
In 2020, Luet al.have synthesized a diverse array of functionalizedgem–difluoroalkene-containing alcohol compounds 97 through the nickel-catalyzed allylic defluorinative reductive cross-coupling ofα-(trifluoromethyl)styrenes with epoxides 96 (Scheme 38) [68].In some cases, the defluorinative products were obtained as a mixture with addition by-products.Thegem–difluoroalkene 97 was formedviatheβ-F elimination of the key intermediate 99,whereas the by-product 98 was produced by the protonation of the intermediate 99.

Scheme 35.The Rh-catalyzed addition reaction of arylboronic esters or phenylboronic acid to α-(trifluoromethyl)styrenes.
Scheme 36.The Ir-catalyzed coupling reaction of α-(trifluoromethyl)styrene with benzoylformic acid.
Scheme 37.The Cu-catalyzed borylation of (3,3,3-trifluoroprop-1-en-2-yl)benzene.
Scheme 38.The Ni-catalyzed reductive couplings of α-(trifluoromethyl)styrene with epoxides.
In the same year, Wanget al.successfully synthesized a variety ofgem–difluorobishomoallylic alcohols 101 fromα-(trifluoromethyl)styrenes 9 and epoxides 100viaa titanocenecatalyzed reductive domino reaction.The by-product, CF3-containing alcohol 102, was also detected (Scheme 39) [69].
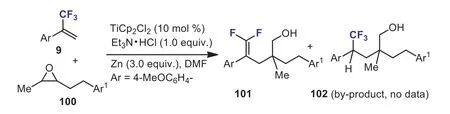
Scheme 39.The Ti-catalyzed reductive couplings of α-(trifluoromethyl)styrene with epoxides.
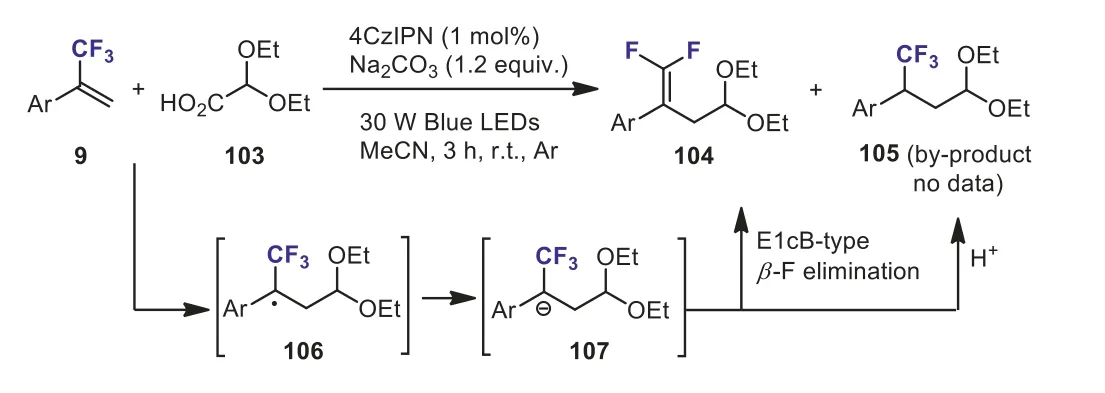
Scheme 40.Organo-photoredox catalyzed defluoroacetalation of α-trifluoromethyl alkenes with glyoxylic acid acetal.
In 2020, Yi and Weiet al.developed a mild and transitionmetal-free organo-photoredox catalyzed defluoroacetalation ofαtrifluoromethyl alkenes 9 with glyoxylic acid acetal 103.This protocol was a straightforward access to diverse maskedγ,γdifluoroallylic ketones 104.The plausible reaction mechanism involved the formation of the stableα-CF3benzyl radical 106 and the sp3-hybridized carbanion intermediate 107.The intermediate 107 underwentβ-fluorine elimination to afford the desired product 104.However, as for someα-trifluoromethyl alkenes having electron-withdrawing group, the carbanionic intermediate 107 could be protonated, giving the competitive hydro-acetalation byproduct 105 (Scheme 40) [70].
Recently, Zhouet al.described an novel visible-light-drivenNradical-mediated tandem radical cyclization/defluorinated alkylation ofα-(trifluoromethyl)styrenes withβ,γ-unsaturated hydrazones (Scheme 41) [71].This method provided an alternative approach to valuable dihydropyrazole-fusedgem–difluoroalkenes under redox-neutral, metal-free, and mild conditions.However, when the reaction ofα-(trifluoromethyl)styrene withβ,γunsaturated hydrazone 108 was performed in the presence of water (CH3CN:H2O = 4:1), an inseparable mixture ofgem–difluoroalkene 109 and hydroaddition product 110 were formed.The ratio of 109 and 110 was 2:1.The authors suggested that the radical intermediate 111 and the carbanion intermediate 112 might be formed during this photocatalytic cycle.The carbanion intermediate 112 could be transformed to CF3-containing product 110 by trapping a proton from water.
In 2021, Wanget al.developed a chromium-catalyzed allylic defluorinative ketyl olefin coupling of aldehydes 113 withα-(trifluoromethyl)styrenes.This novel method enabled the rapid synthesis of diversegem–difluorohomoallylic alcohols under mild and reductive conditions.The authors found that whenα-(trifluoromethyl)styrenes bearing a strong electron-withdrawing group were subjected to the optimized reaction conditions, in addition to the desired allylic defluorinative product 114, a small amount of the nondefluorination by-product 115 was also detected(Scheme 42) [72].
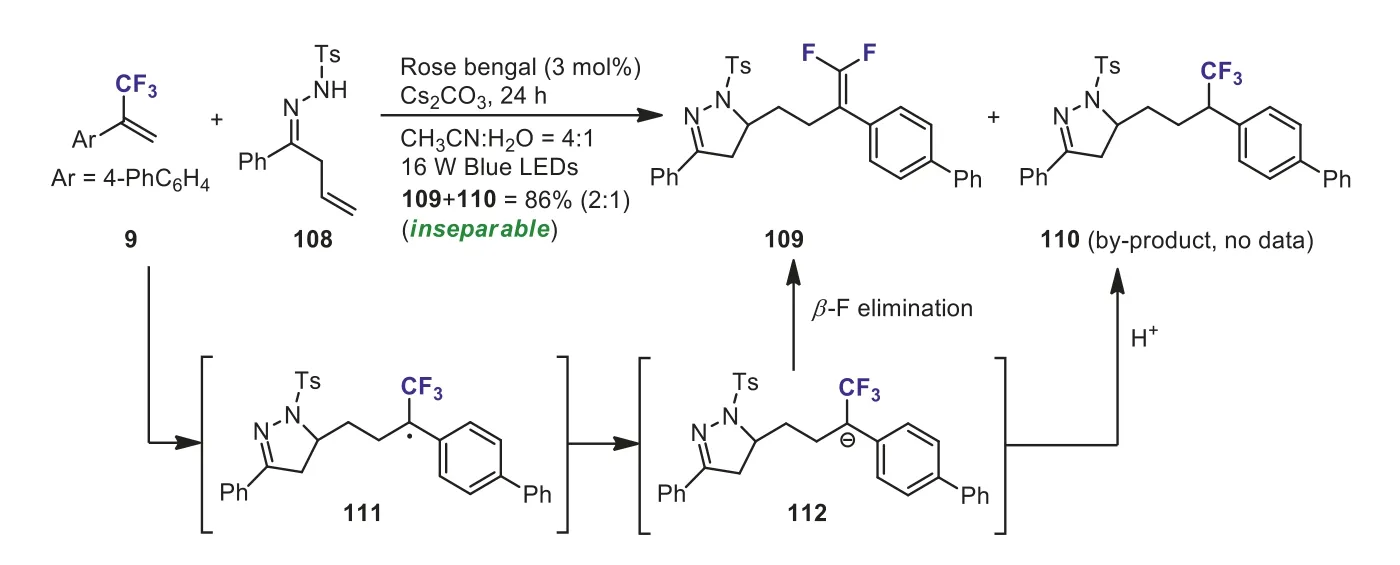
Scheme 41.Visible light-induced photoredox catalytic coupling of α-(trifluoromethyl)styrene with β,γ-unsaturated hydrazone.
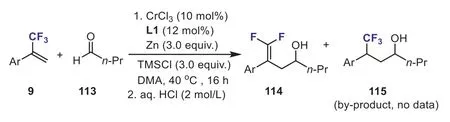
Scheme 42.The Cr-catalyzed coupling reaction of α-(trifluoromethyl)styrene with aldehyde.
5.Conclusions
In summary, this review highlights the recent advances in cycloaddition and hydroaddition reaction ofα-(trifluoromethyl)styrenes with various substrates without defluorination.Although several elegant transformations ofα-(trifluoromethyl)styrenes to CF3-containing compounds have been reported in the past two decades, synthetic application of this strategy is still in its infancy mainly because of the simultaneous formation of the defluorination products, which are often the main reaction products.Despite the fact that the yields of hydroaddition products are generally low in most cases, plenty of room for improvement still remains with regard to reaction types, yields and substrate scope.The critical challenge is to suppress the undesired defluorination reaction.It is anticipated that the hydroaddition reaction ofα-(trifluoromethyl)styrenes without affecting CF3group will be an alternative approach to valuable CF3-containing compounds.We hope that this short review about the rarely explored hydroaddition reaction ofα-(trifluoromethyl)styrenes would arise great interest in the chemical community and help to obtain deep insight into hydroaddition reaction.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
We are grateful for the financial support from the National Natural Science Foundation of China (No.21472043).
 Chinese Chemical Letters2022年5期
Chinese Chemical Letters2022年5期
- Chinese Chemical Letters的其它文章
- Recent advances in enhancing reactive oxygen species based chemodynamic therapy
- An integrative review on the applications of 3D printing in the field of in vitro diagnostics
- Recent developments of droplets-based microfluidics for bacterial analysis
- Dynamics and biological relevance of epigenetic N6-methyladenine DNA modification in eukaryotic cells
- Recent progress in advanced core-shell metal-based catalysts for electrochemical carbon dioxide reduction
- Recent advances in carbon-based materials for electrochemical CO2 reduction reaction