
基于生物信息学分析揭示circRNA-m iRNA-m RNA调控网络在胃癌中的作用
2022-06-18 08:22王艳君许鑫鑫鲁意迅王风王洁
河南医学研究 2022年11期
王艳君,许鑫鑫,鲁意迅,王风,王洁
(1.郑州大学附属儿童医院/河南省儿童医院 外科重症监护室,河南 郑州 450000;2.中国人民解放军总医院第一医学中心 普通外科,北京 100853)
胃癌是世界范围内最常见的恶性肿瘤之一,其相 关病死率在全球排第4位,中国是胃癌大国,每年新增病例约67万人,占全世界的40%[1]。近年来,以新的诊断标志物[2]、微创手术[3]、靶向治疗[4]和免疫治疗[5]为代表,胃癌的诊治取得了巨大进展。然而,我国进展期胃癌患者的5 a生存率仍不足30%[6]。因此,迫切需要寻找有效的生物标志物和可能的治疗靶点,以改善胃癌的临床预后。环状RNA(circular RNA,circRNA)是一类无5’端帽子和3’端尾巴的闭合环状RNA分子[7]。虽然circRNA分子40 a前已被发现,但其功能直到最近才逐渐被揭示[8]。越来越多的研究表明,circRNA参与各类恶性肿瘤的发生和进展,包括肺癌、乳腺癌、肝癌、胃癌等[9],使之成为非编码RNA(non-coding RNA,ncRNA)家族中一颗冉冉升起的“新星”。与线性RNA相比,circRNA的稳定性[10]、保守性[11]和组织器官特异性[12]赋予了其作为新的诊断标志物以及治疗靶点的巨大潜力。截至目前,一些研究已初步探索了circRNA在胃癌发生、发展中的作用和机制,其中研究最多的机制是它可以充当miRNA的“分子海绵”。例如,circLMTK2通过充当miR-150-5p的分子海绵来促进胃癌的增殖和转移[13]。同样,circHIPK3通过吸附miR-637和调节下游基因AKT1进而促进胃癌的增殖和转移[14]。此外,circREPS2通过充当miR-558的分子海绵和调节RUNX3/β-catenin来抑制胃癌的进展[15]。然而,绝大部分circRNA在胃癌中的生物学功能和作用机制尚不清楚。本研究从GEO数据库中获取了3个circRNA表达数据集(GSE78092、GSE83521、GSE93541),通过对比胃癌和癌旁组织,鉴定了两条差异表达的cirRNA(differentially expressed circRNA,DE circRNA),利用生物信息学方法对circRNA-miRNA和miRNA-mRNA进行靶向预测,并进行了GO和KEGG富集分析,最终筛选出10个核心基因并建立了circRNA-miRNAmRNA/核心基因的分子调控网络。
1 资料与方法
1.1 资料GEO数据库(http://www.ncbi.nlm.nih.gov/geo)包含来自世界各国的大量高通量基因组表达数据。本研究从GEO数据库中选取了3个数据集(GSE78092、GSE83521、GSE93541),这些数据集都是使用基因芯片或二代测序技术检测胃癌和癌旁组织中circRNA分子的表达量。
1.2 研究方法
1.2.1 DE circRNA分子的确定 对3个数据集的原始数据进行标准化及log2转换。使用R 软件中的Limma包筛选胃癌和癌旁组织中的DE circRNA。截断值设定为|log2Fold Change|>1且P<0.05。将3个数据集取交集后筛选出的DE circRNA视为候选circRNA。使用R软件中ggplot2和维恩图VENNY 2.1(https://bioinfogp.cnb.csic.es/tools/venny/index.html)进行数据可视化。
1.2.2 候选miRNA的预测 同时使用4个生物信息数据库来研究DE circRNA和miRNA之间的潜在靶向关系,包括CircInteractome(https://circinteractome.nia.nih.gov/)、circBank(http://www.circbank.cn/index.html)、starBase v2.0(http://starbase.sysu.edu.cn/index.php)以及circAtlas 2.0(http://circatlas.biols.ac.cn/)。能够同时被3个或3个以上数据库预测到的miRNA视为DE circRNA的候选miRNA。
1.2.3 miRNA下游靶基因的预测 同时使用TargetScan(http://www.targetscan.org/vert_72/)、miRDB(http://mirdb.org/)以 及 miRwalk 3.0(http://mirwalk.umm.uni-heidelberg.de/)数据库预测miRNA-mRNA之间的可能的靶向关系。至少在2个数据库中预测到的靶基因被认定为miRNA的潜在靶点。
1.2.4 GO和KEGG富集分析 本研究使用DAVID数据库v6.8(https://david.ncifcrf.gov/)进行靶基因的GO[16]和KEGG[17]富集分析。GO数据库(http://www.geneontology.org)可以为基因组数据提供功能注释,包括生物学过程(biological processes,BP)、细胞组份(cellular component,CC)以及分子功能(molecular function,MF)等[16]。KEGG 数 据 库(http://www.genome.ad.jp/kegg/)可以为基因提供系统的分析、注释和可视化[17]。使用R软件中ggplot2包进行数据可视化。截断值设定为P<0.05。
1.2.5 核心基因识别及模块化分析 使用STRING数据库(https://string-db.org/)预测蛋白质-蛋白质互作关系[18](截断值设定为信度评分>0.9分),使用Cytoscape v3.7.2软件[19]对蛋白质-蛋白质互作网络(protein-protein interaction,PPI)进行可视化分析。利用cytohubba插件依据节点等级识别核心基因,使用MOCODE插件筛选不同的PPI模块。参数设置如下:degree cutoff=2,node score cutoff=0.2,k-core=2,maximum depth=100。
1.2.6 宿主基因及核心基因生存分析 应用Kaplan-Meier plotter在线数据库(http://kmplot.com),依据875例胃癌患者的现有数据,分析宿主/核心基因与总生存期(overall survival,OS)之间的相关性[20]。统计分析高表达与低表达宿主/核心基因患者的生存差异。P<0.05为差异有统计学意义。
1.2.7 circRNA-miRNA-mRNA调控网络的建立 将核心基因与每个miRNA的靶基因取交集,获得miRNA-核心基因配对关系。基于circRNA-miRNA和miRNA -核心基因之间的靶向关系,建立circRNA-miRNA -核心基因的子网络。使用Cytoscape v3.7.2软件对竞争性内源RNA(competing endogenous,ceRNA)调控网络进行数据可视化。
2 结果
2.1 胃癌中DE circRNA的鉴定GSE78092数据集中共筛选出199个DE circRNA,其中上调53个,下调146个。GSE83521数据集中共筛选出150个DE circRNA,其中上调80个,下调70个。GSE93541数据集中共筛选出418个DE circRNA,其中上调216个,下调202个。将这些DE circRNA取交集,得到2个候选的circRNA:hsa_circ_0001658和hsa_circ_0032683。Kaplan-Meier Plotter分析显示,ARID1B(hsa_circ_0001658的宿主基因)和NEK9(hsa_circ_0032683的宿主基因)与胃癌患者的预后有相关性。见表1。

表1 候选DE circRNA的基本信息
2.2 预测circRNA-miRNA和miRNA-mRNA靶向关系基于CircInteractome、circBank、starBase和circAtlas数据库,鉴定出7个目标miRNA。基于TargetScan、miRDB和miRwalk 3.0数据库获得miRNA的靶mRNA,共预测出1 796对miRNA-mRNA。将miRNA的靶基因汇聚在一起并去重后,获得1 695个靶基因。见表2。
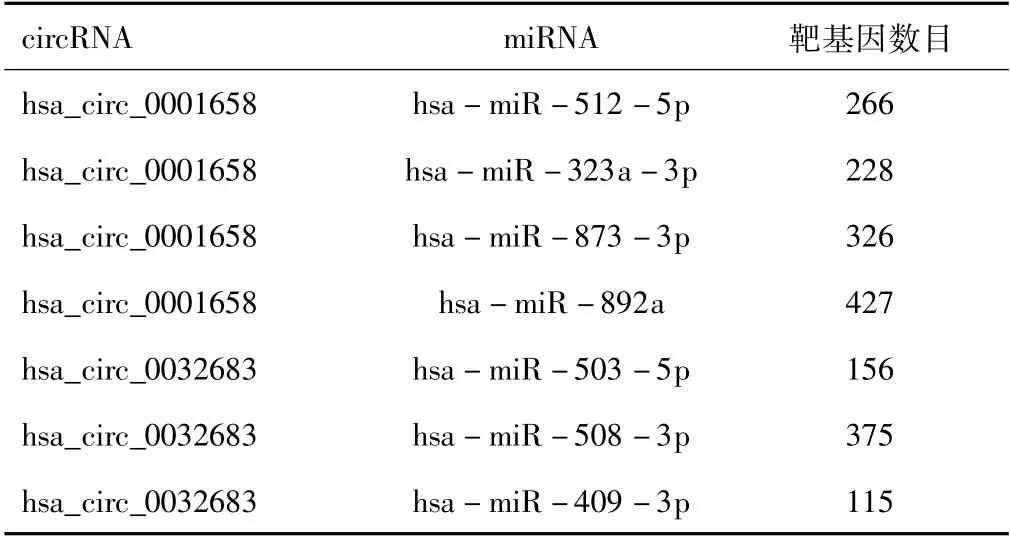
表2 circRNA-miRNA以及miRNA-mRNA靶向关系预测结果
2.3 GO和KEGG富集分析将上述1 695个靶基因通过DAVID数据库进行GO和KEGG富集分析。GO富集分析结果显示,CC注释中,靶基因最常见的细胞定位是细胞核,其次是细胞质和核质等。BP注释中,靶基因主要参与模板DNA的转录、转录调控、针对RNA聚合酶Ⅱ启动子的转录调控及信号转导、细胞增殖调控、蛋白质磷酸化、细胞黏附等。靶基因的MF主要是参与蛋白质、金属离子、DNA、ATP、锌离子、转录因子、特异序列DNA、poly(A)RNA、钙离子等的结合以及蛋白质异源二聚体活动等。根据KEGG富集分析结果,上述靶基因主要富集在癌症相关通路、PI3K-Akt信号通路、MAPK信号通路、HTLV-I感染、cAMP信号通路、actin细胞骨架调控、Ras信号通路、泛素介导的蛋白质降解、癌症中的蛋白聚糖依赖途径、Rap1信号通路等。
2.4 核心基因的识别和模块化分析通过STRING数据库和Cytoscape(cytohubba插件),共筛选出10个核心基因:CTNNB1、MAPK1、KRAS、APP、FN1、CDH1、CASP3、BTRC、UBA52、CREB1。见表3。

表3 核心基因的详细信息
2.5 核心基因的生存分析Kaplan-Meier Plotter分析结果显示,8个核心基因(CTNNB1、MAPK1、KRAS、APP、FN1、CDH1、CASP3、BTRC)对胃癌患者的OS有影响,而核心基因UBA52和CREB1对胃癌患者OS的影响无统计学意义(P>0.05)。
2.6 circRNA-miRNA-mRNA/核心基因调控网络的构建最终确定了2个DE circRNA、7个miRNA和10个核心基因,建立了circRNA-miRNA-mRNA/核心基因的子网络,以此来显示胃癌中最有可能的调控网络。见图1。
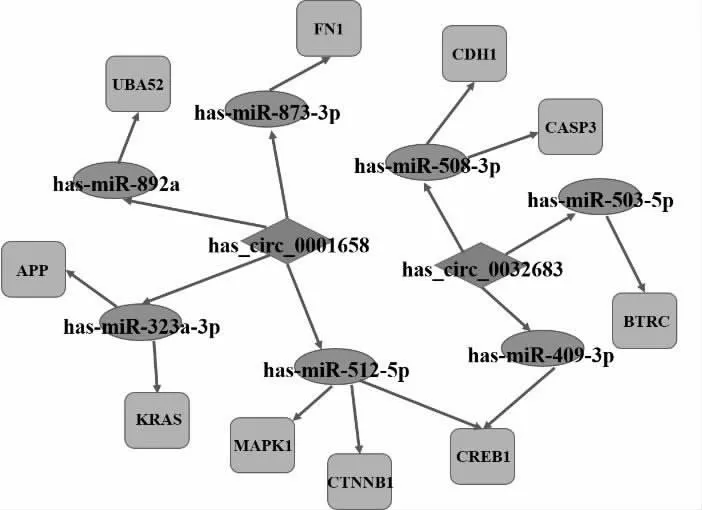
图1 以circRNA-miRNA-mRNA/核心基因相互作用关系构建ceRNA调控网络
3 讨论
近年来,胃癌的诊断和治疗取得了巨大进展,但胃癌的预后依然不佳。传统生物标志物(如癌胚抗原、糖类抗原199、糖类抗原125等)的诊断效能较差[21],新型特异性生物标志物缺乏,导致胃癌的早期诊断率较低,加之有效治疗靶点的缺乏,严重制约了胃癌的治疗效果。因此,鉴定特异、高效的胃癌诊断标志物及潜在的治疗靶点具有重要意义。过去几十年间,高通量测序技术和生物信息学的迅猛发展,为多种疾病特别是肿瘤提供了新的研究工具和方法。基于生物信息学方法筛选胃癌相关基因、寻找新的诊断标志物和治疗靶点已成为可能。本研究以circRNA为研究对象,旨在通过全面的生物信息学分析,识别胃癌相关circRNA并研究其可能的调控网络。
首先,本研究鉴定了2个胃癌相关DE circRNA:hsa_circ_0001658和hsa_circ_0032683。生存分析显示,ARID1B(hsa_circ_0001658的宿主基因)和NEK9(hsa_circ_0032683的宿主基因)均与胃癌患者的OS相关。这些发现表明,分别由ARID1B和NEK9生成的hsa_circ_0001658和hsa_circ_0032683可能参与了胃癌的恶性生物学行为。最近的一项研究表明,hsa_circ_0001658可通过hsa_circ_0001658/miR-375/PAX6轴参与胃癌进展[22]。此外,另一项研究证实hsa_circ_0001658可通过miR-382-5p/YB-1轴促进骨肉瘤细胞的增殖和转移[23]。而针对hsa_circ_0032683(circNEK9)的研究,Yu等[24]发现circNEK9可以通过miR-409-3p/MAP7轴加快胃癌的进展,这与本研究预测结果相符。
本研究通过生物信息学分析筛选出匹配概率最高的circRNA-miRNA和miRNA-mRNA配对关系,对miRNA靶基因进行GO和KEGG富集分析,结果表明,在BP注释中,靶基因主要涉及转录调控、信号转导、细胞增殖、细胞黏附等肿瘤的恶性行为。此外,KEGG富集分析还显示,靶基因在肿瘤相关通路中明显富集。从PPI网络中筛选出10个核心基因:CTNNB1、MAPK1、KRAS、APP、FN1、CDH1、CASP3、BTRC、UBA52、CREB1。生存分析提示核心基因CTNNB1、MAPK1、KRAS、APP、FN1、CDH1、CASP3、BTRC对胃癌患者OS有影响,而核心基因UBA52、CREB1对胃癌患者OS无影响。例如,在这10个核心基因中,由CTNNB1基因编码的β-catenin是WNT/β-catenin通路的关键组成部分,它参与了包括胃癌在内的多种癌症的进展。研究表明,WNT/β-catenin通路的异常激活可以促进胃癌的进展[25]。KRAS是一个众所周知的致癌基因,最近的研究表明KRAS激活可促进胃癌细胞的上皮-间质转化(epithelial-to-mesenchymal transition,EMT)和转移[26]。FN1基因编码纤维连接蛋白1,它可以调节细胞黏附、迁移、增殖、分化和肿瘤转移等生物学过程。有研究表明,FN1在胃癌组织中的表达上调,且与胃癌的不良预后显著相关[27]。CDH1编码E-cadherin,这是维持细胞黏附的关键。E-cadherin低表达可以诱导EMT,而EMT正是癌细胞获得迁移和侵袭能力的原因之一[28]。E-cadherin在胃癌中有抑癌作用,CDH1突变与遗传性弥漫性胃癌的发生密切相关[29]。UBA52在胃癌中的作用仍然存在争议,Tian等[30]通过PPI网络和生物信息学分析证实UBA52与胃癌的进展和转移密切相关,另一项研究表明UBA52在结肠癌中表达上调,而在胃癌中的表达无显著变化[31]。有研究显示,CREB1的下调可抑制胃癌细胞体内和体外生长[32]。虽然UBA52及CREB1对患者的OS无显著影响,但二者在胃癌中的作用仍待进一步探讨。综上,本研究发现的核心基因绝大部分都与胃癌的恶性行为有关,包括肿瘤的发生、侵袭、迁移、转移、EMT等,这与本研究中GO和KEGG富集分析的结果一致。
最后,本研究建立了circRNA-miRNA-mRNA/核心基因的ceRNA调控网络,确定了核心基因及其调控网络,有望为胃癌诊断提供潜在的生物标志物或治疗靶点,拓展了circRNA通过ceRNA网络调控胃癌发生、发展的分子机制。然而,本研究的局限性在于缺乏循证医学证据,数据分析结果也需要细胞和动物实验进一步验证。
本研究确定了2个胃癌相关circRNA和10个核心基因,并建立了circRNA-miRNA-mRNA/核心基因的ceRNA调控网络。通过识别DE circRNA进而构建ceRNA网络,可能为胃癌的诊断提供新的生物标志物或潜在的治疗靶点。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
健康体检与管理(2022年2期)2022-04-15
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年3期)2021-12-03
昆明医科大学学报(2021年5期)2021-07-22
天津医科大学学报(2021年2期)2021-03-29
中国生殖健康(2020年7期)2020-12-10
腹腔镜外科杂志(2016年10期)2016-06-01
医学研究杂志(2015年9期)2015-07-01
医学研究杂志(2015年7期)2015-06-22
