
Genome-Wide Analysis of von Willebrand Factor A Gene Family in Rice for Its Role in Imparting Biotic Stress Resistance with Emphasis on Rice Blast Disease
2022-06-16 11:38SuhasGorakhKarkuteVisheshKumarMohdTasleemDwijeshChandraMishraKrishnaKumarChaturvediAnilRaiAmithaMithraSevanthiKishorGaikwadTilakRajSharmaAmolkumarSolanke
Rice Science 2022年4期
Suhas Gorakh Karkute, Vishesh Kumar, Mohd Tasleem, Dwijesh Chandra Mishra, Krishna Kumar Chaturvedi, Anil Rai, Amitha Mithra Sevanthi, Kishor Gaikwad, Tilak Raj Sharma, Amolkumar U. Solanke
Research Paper
Genome-Wide Analysis of von Willebrand Factor A Gene Family in Rice for Its Role in Imparting Biotic Stress Resistance with Emphasis on Rice Blast Disease
Suhas Gorakh Karkute1, Vishesh Kumar1, Mohd Tasleem1, Dwijesh Chandra Mishra2, Krishna Kumar Chaturvedi2, Anil Rai2, Amitha Mithra Sevanthi1, Kishor Gaikwad1, Tilak Raj Sharma3, Amolkumar U. Solanke1
(Indian Council of Agricultural Research (ICAR)-National Institute for Plant Biotechnology, New Delhi 110012, India; ICAR-Indian Agricultural Statistics Research Institute, New Delhi 110012, India; ICAR-Division of Crop Science, New Delhi 110001, India)
von Willebrand factor A (vWA) genes are well characterized in humans except for few BONZAI genes, but the vWA genes are least explored in plants. Considering the novelty and vital role of vWA genes, this study aimed at characterization of vWA superfamily in rice. Rice genome was found to have 40 vWA genes distributed across all the 12 chromosomes, and 20 of the 40 vWA genes were unique while the remaining shared large fragment similarities with each other, indicating gene duplication. In addition to vWA domain, vWA proteins possess other different motifs or domains, such as ubiquitin interacting motif in protein degradation pathway, and RING finger in protein-protein interaction. Expression analysis of vWA genes in available expression data suggested that they probably function in biotic and abiotic stress responses including hormonal response and signaling. The frequency of transposon elements in the entire 3K rice germplasm was negligible except for 9 vWA genes, indicating the importance of these genes in rice. Structural and functional diversities showed that the vWA genes in a blast-resistant rice variety Tetep had huge variations compared to blast-susceptible rice varieties HP2216 and Nipponbare. qRT-PCR analysis of vWA genes ininfected rice tissues indicated,,andas the optimal candidate genes for disease resistance. This is the first attempt to characterize vWA gene family in plant species.
von Willebrand factor A; biotic stress; abiotic stress; rice blast disease;
Proteins containing von Willebrand factor A (vWA) domain are present in almost all the organisms belonging to eukaryote, prokaryote and archaea (Whittaker and Hynes, 2002). Many of these proteins are cell adhesion and extracellular matrix proteins (Tuckwell, 1999). The vWA protein in humans is a large multimeric glycoprotein that contains the vWA domain, and is very well characterized because of its role in hemostasis by mediating platelet adhesion to the site of vascular injury (Reininger, 2008). The protein functions as a multimer which is formed by-glycosylation of monomers into dimers and subsequently into multimers. Mutations in vWA genes result in von Willebrand factor disease in humans where the individual suffers from defects in blood clotting (Bharati and Prashanth, 2011). The vWA domain has a typical α/β Rossmann-fold involving in alternating α-helices and β-strands, and has a metal ion-dependent adhesion site, which plays an important role in ligand binding (Whittaker and Hynes, 2002). The vWA domain is primarily involved in protein- protein interactions in multiprotein complexes.
Unlike in humans, vWA domain-containing proteins in plants are not well characterized. To date, only copine proteins which contain vWA domain and two calcium-dependent phospholipid-binding C2 domains inand rice are characterized to some extent.genome has three copine genes (,and) performing overlapping function as negative regulators of programmed cell death and defense response (Liu et al, 2005; Yang et al, 2006). Two orthologs of copine genes in rice,and, function in a similar way by suppressing broad-spectrum disease resistance (Yin et al, 2018). Additionally, vWA proteins, having really interesting new gene (RING) finger domain specific to plants likeand rice, have been identified (Whittaker and Hynes, 2002). Recently, two vWA genes have been identified in gene expression data in response to biotic stress in rice. One is up-regulated in gall midge resistant rice genotype after gall midge infection (Rawat et al, 2012), whereas the other has higher expression ininfected rice plants, indicating its role in parasitic weed resistance (Swarbrick et al, 2008). Interestingly, these two genes were also found to be highly up-regulated in response toinfection in an internationally well-known blast-resistant rice variety Tetep (Kumar et al, 2021). Previously, fine mapping of panicle blast resistance genein rice showed the presence of these two genes in the locus, suggesting their roles in blast resistance (Fang et al, 2019). All these reports fromand rice indicate that vWA genes can be crucial players in imparting biotic stress resistance. However, there is no single exploratory or systematic study on these genes available in plant species. Limited information is available on the remaining two copine genes and no molecular or functional information is available for any other vWA proteins in plants. Considering the importance of these proteins in biotic stress response, it is necessary to identify and characterize the vWA protein family in plants.
Rice (L.) is the most important crop for global food security as it feeds more than half of the world’s population. The availabilities of whole- genome sequence information and various molecular biology tools in rice make it a model crop system for study. Moreover, rice yield is severely affected by different biotic stresses like bacterial blight, sheath blight and rice blast. After identification of more than 100 resistance (R) genes governing blast resistance in rice, the focus has now been shifted towards the identification of defense regulator (DR) genes (Li et al, 2019). Several DR genes govern broad-spectrum durable blast resistance, and hence it is worthwhile if more such genes are identified. vWA genes are known to be up-regulated during biotic stress response in rice and may work as broad-spectrum DR genes. Therefore, in the present study, genome-wide identification and characterization of vWA family proteins are carried out to understand their structural and functional diversities, and their response to a representative biotic stress (rice blast infection). We have mined the whole rice genome to identify vWA genes based on vWA domain sequence. All these genes were analyzed for evolutionary relationship and explored for their expression profiles from Genevestigator and RiceMetaSys databases. Expression analysis of selected genes was carried out by qRT-PCR to identify their responses toinfection in rice. Owing to the diversities in domains present in the vWA proteins, we have also detected their expression profiling in abiotic stress to identify stress responsive genes.
RESULTS
Chromosomal distribution and structural characterization of vWA domain-containing genes
Complete rice genome scan found a total of 40 genes that possessed at least one vWA domain. Although some of these genes have already been characterized and named, for more convenience, all these OsvWA genes were renamed asto(Table S1), and were distributed on all the 12 chromosomes of rice (Fig. 1). The maximum number (6) of genes was clustered on chromosome 11 includingandgenes that are reported across several biotic stress responses. Chromosomes 3 and 6 both had five OsvWA genes. In contrast, chromosomes 5, 7 and 9 had only one vWA gene each.() on chromosome 5 is a negative regulator of fungal diseases, and() on chromosome 9 is expressed in response to both biotic and abiotic stresses (Kazan, 2017). The smallest genecodes for a copine protein of 387 amino acid residues, whereas the largest onecodes for phytochrome and flowering time 1 (PFT1) protein of 842 amino acid residues.
The presence of vWA domain in the proteins was further confirmed by subjecting them to the SMART tool. Structural analysis of all the protein sequences provided important information about various kinds of domains that were present in these proteins along with the vWA domain (Table S1). These domains included RING finger, second conserved domain of protein kinase C (C2), ubiquitin interacting motif (UIM), ATPases associated with various cellular activities (AAA), Transcription factor IIH (TFIIH) C1-like domain (C1_4), and structural classification of proteins (SCOP) d1bg1a1 (Fig. S1). Moreover, information of subcellular localization of each vWA protein in different locations, such as cytoplasm, chloroplast, mitochondria, endoplasmic reticulum, nucleus, and plasma membrane, is listed in Table S1, which supported its diverse roles.
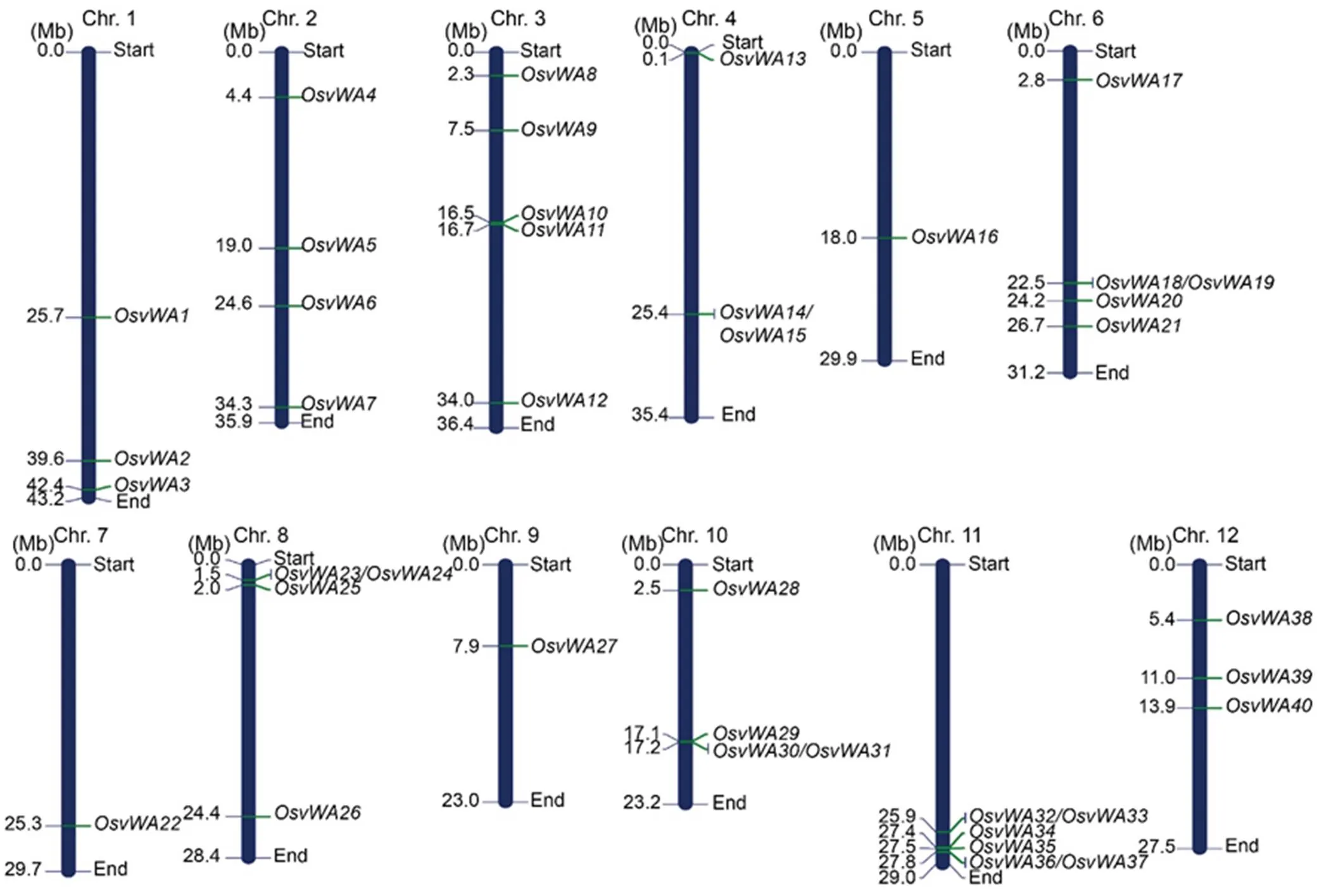
Fig. 1. Genome wide distribution of vWA gene family on 12 rice chromosomes.
The presence of transposon elements (TEs) in the vWA family genes in 3K rice genotypes by Rice Transposon Insertion Polymorphism (RTRIP) database revealed that many genes harbor transposon insertions in the coding domains (CDS), intron, upstream or downstream sequence, or untranslated region (Table S2). The maximum TEs were located in the non- coding regions, and therefore may not affect the normal functions of the genes. However, TEs in a few genes, such as,,,,,and, were located in CDS region, and may affect the functions of the gene in those particular genotypes. Among them,andwere the only two genes having two transposon insertions each in the CDS region. Further,showed as many as eight TE insertions, which is the maximum among the family. However, the frequency of these elements in the entire 3K rice germplasm was negligible except for 9 of the 40 vWA genes, indicating the importance of the function of these genes in rice.
Single nucleotide polymorphisms (SNPs) were identified for all the vWA genes in Tetep (blast resistant variety) and HP2216 (blast susceptible variety) using Nipponbare (blast susceptible variety) sequence as the reference (Table S3). The sequences of the genes in HP2216 were found to be highly similar to those in Nipponbare, with many genes having no SNPs or having very few SNPs. In contrast, the sequences of the vWA family genes in Tetep were highly variable with a large number of SNPs except for very few genes. Onlywas completely conserved in all the three genotypes with no SNPs.
Phylogenetic analysis and evolutionary relationship
An unrooted phylogenetic tree of the 40 OsvWA protein sequenceswas generated for a deeper understanding of their phylogenetic relationship during evolution (Fig. 2-A). The phylogenetic tree revealed two main clades (Clade I and Clade II), and furthermore divided into six groups (G1 to G6), where the largest group (G1) had 14proteins and the smallest group (G3) had only 2. The evolutionary relationship among the OsvWA genes by the Circos analysis showed large events of duplication of gene segments (Fig. S2). As many as 20 members of the OsvWA family were unique and they did not share any similarities with other members. The genes which shared a large part of the nucleotide sequence similarity have probably originated from a single gene and following gene duplication accumulated changes in nucleotide sequences during evolution (Yu et al, 2005). Interestingly,andwhich are on chromosome 11,have large DNA fragments in common withandwhich are on chromosome 6, i.e., the cDNA ofis 90% similar to, whereas that ofis 86% similar to.
To identify common regulatory elements in promoter sequences of the vWA genes, phylogenetic analysis was performed in Tetep and Nipponbare. The phylogeny revealed that the promoter sequences of corresponding genes in Tetep and Nipponbare were similar with minor variations (Fig. 2-B). However, between the two varieties, corresponding promoter sequences of,,andgenes had large variations. In each variety, promoter sequences ofandwere found to be similar toand, respectively, whose coding sequences were also closely related.
Expression profiling of OsvWA genes
Analysis by Genevestigator at different developmental stages of rice showed significant differential expression of all OsvWA genes at all developmental stages (Fig. S3). Expression of,,andwas highat the reproductive stages like grain filling and ripening stages. Investigation of gene expression in anatomical parts revealed that all the 40 OsvWA genes were expressed in at least one tissue of rice (Fig. S4). Some of the genes like,,,,andshowed variable expression levels in almost all the tissues. The expression ofhaving zinc finger (C3HC4-type RING finger) domain was highly up-regulated in endosperm but was very meager in other tissues.containing the UIM domain along with vWA domain was highly up-regulated in the primary root tip.was found to be highly expressed in sheath, peduncle and shoot tissues, whereasexpression was in leaf, panicle and shoot tissues.
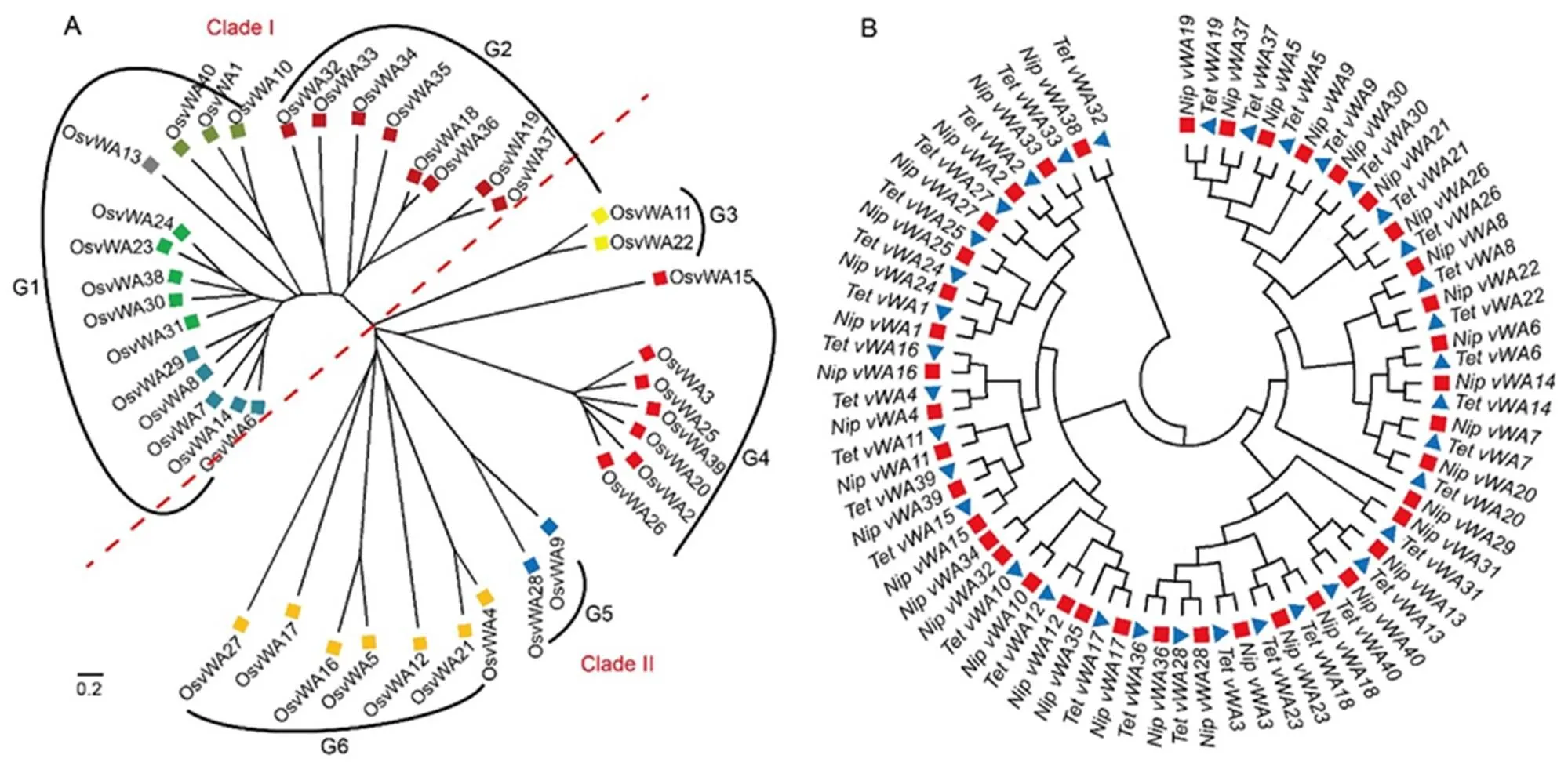
Fig. 2. Evolutionary relationship of vWA proteins (A) and promoter sequence of vWA genes (B) in rice.
Different colored squares in A represent proteins in different subclades. Blue triangle and red square in B represent promoters from blast resistant variety Tetep (Tet) and susceptible variety Nipponbare (Nip), respectively. The evolutionary history was inferred using the neighbor-joining method. The evolutionary distances were computed using the Poisson correction method.
Several OsvWA genes were found to be apparent response to various biotic stresses (Fig. S5).,andwere highly up-regulated whereaswas down-regulated in response toat all the stages from 12 h post inoculation (hpi) to 72 hpi. Similarly,,,,,andgot induced in response to the infection ofpv., however, infection with different strains ofpv.caused down-regulation of,andgenes. Therefore, these three genes showed a differential response to different strains of.. Interestingly, the expression ofwas highly down-regulated at all the stages after infection of both strains ofand.
Bothandshowed higher expression in rice tissues after treated with insect(gall midge), parasitic weedand infection ofpv.. Based onthe RiceMetaSysB database, the expression of,,,andgenes in leaf tissues andandgenes in root tissues afterinfection showed significant induction (Table S4). In addition, the expression of,andwas slightly induced after bacterial blight infection. Except for, the other three genes belonged to the same group of copine genes, known to play a role in bacterial disease response in plants.
OsvWA genes responsive to abiotic stress like drought and salt were also identified from the RiceMetaSys database.,,andwere significantly up-regulated during drought stress, whereasgot down-regulated in drought- stressed plants. Interestingly, the RiceMetaSys data showed drastic up-regulation (125-fold) ofgene under salt stress at the vegetative stage of rice, making it play roles in both biotic and abiotic stress responses and a point of cross-talk.
In addition, there were a few genes showing significant responses to chemical stress (Fig. S5).got up-regulated in root samples after exposure to aluminum (Al), arsenate (As), and cadmium (Cd) stresses, while,andgot down-regulated by As, lead (Pb), Cd and dexamethasone treatments in rice. Two other genesandgot down-regulated only under As and Pb stresses. Further, some of the genes were also identified as light-responsive genes.was the only one that showed higher expression in the dark, whereas,,,andwere all down-regulated in the dark in rice.
andgenes showed up-regulation in rice seedlings after 3 h of salicylic acid application. Similarly, cytokinin treatment also induced the expression ofin both leaf and root tissues, andonly in the root.andin rice seedlings showed up-regulation after gibberellin (GA3), kinetin and naphthylaceticacid (NAA) treated.andwhich showed enhanced expression under biotic stress were significant down- regulation after GA3 and NAA treatments.
MapMan tool was used to identify the metabolic pathways of biotic stress response wherein the OsvWA genes were involved. OsvWA genes were found to be involved mainly in three pathways: proteolysis, signaling and pathogenesis related (PR) proteins with many genes falling in the proteolysis process (Fig. S6).
Expression analysis of selected OsvWA genes during M. oryzae infection
Panicles of Tetep and HP2216 inoculated withshowed differential disease response to infection. At 48 hpi, Tetep showed no symptoms of disease while HP2216 started showing small lesions at the inoculated sites, and panicles at 48 hpi were used for expression analysis of the representative 22 members of the vWA family selected based on phylogenetic analysis, Genevestigator analysis and domain informationof protein. Further, the subsequent observations clearlydistinguished the resistant and susceptible characteristics of the two varieties to blast disease. Lesions of HP2216 grew in size during the progression of the disease but Tetep showed symptoms of immune response.
infection resulted in induced expression of different gene sets in Tetep and HP2216, indicating the genotype-specific disease response (Fig. 3).,,,,,andgenes in Tetep were highly induced while down-regulated or slightly up-regulated in HP2216 at 48 hpi. On the other hand,,,,,andshowed up-regulation in HP2216 as compared to Tetep. Interestingly,which is the homolog ofwas down-regulated in Tetep but up- regulated in HP2216. We further observed thatandshowed no expression in rice tissues. At 48 hpi, several genes involved in the proteolysis process were up-regulated in Tetep as compared to HP2216. Only one gene,, which is involved in signaling,is earlier characterized asin rice and is known to be involved in biotic stress response (Yin et al, 2018).

Fig. 3. Relative expression analysis of OsvWA genes in rice panicle tissues of Tetep and HP2216 genotypes inoculated withat 48 h post inoculation using qRT-PCR.
Candidate vWA genes for biotic stress response in rice
To narrow down the vWA genes crucial for biotic stress resistance, we compared gene expression profiles of all the vWA genes across the economically important disease infections like blast, sheath blight and bacterial blight. Expression data of qRT-PCR for blast and Genevestigator and RiceMetaSysB for sheath blight and bacterial blight showed several genes expressed in response to these diseases. Thus, we identified 12 vWA genes that were induced in response to biotic stress (Fig. 4), with nine to blast, three to sheath blight and eight to bacterial blight, among which 2 vWA genes (and) were in response to blast, sheath blight and bacterial blight at the same time.
DISCUSSION
vWA genes in humans are critical for blood hemostasis and therefore, they are very well characterized. Contrastingly, in plants, only a few vWA genes such asandinare studied to a limited extent (Yang et al, 2006). From disease responsive transcriptomics in blastresistant and susceptible rice varieties, Kumar et al (2021) identified two novel genes containing vWA domain that are highly induced in resistant variety Tetep. The novelty of the genes, their presence across all the taxons from bacteria to higher eukaryotes, significant roles in humans and scarce information available on such genes in plant species prompted us to characterize the whole gene family in rice, a model species for monocots.

Fig. 4. Venn diagram showing vWA genes expressed in different biotic stresses of blast, sheath blight and bacterial blight.
The 40 vWA family members in rice formed different groups with a variable number of genes, showing diverse evolutionary patterns. Circos analysis showed that 50% of the genes were unique, while the remaining 50% genes had high similarity with one or more members,indicating their common origin. Interestingly, one such pair, i.e.andwith more than 90% sequence similarity in cDNA showed very higher up-regulation level after infection within the resistant variety Tetep. Further,was highly up-regulated after blast infection in Tetep, while its homologwas slightly up-regulated in the susceptible variety HP2216. Thus, such two homologs may provide synergistic effects during the stress response. Moreover, the promoters of these genes were also similar in Tetep and Nipponbare. Promoter sequences of these genes in HP2216 could not be included due to their non-availability in the available databases.() and() genes are closely located on chromosome 11 with a distance of 6.3 kb, whereas their homologs() and() are present on chromosome 6 with a distance of 35 kb. Taking into account the similarity in sequences of promoters and their positions on the chromosomes, there was a tandem duplication of the genomic segment, translocation and subsequent accumulation of mutations in the process of evolution though the functionality has been retained.
Different TEs present in the genic region of the gene play a critical role in the expression and therefore, their analysis is also important. Many of the stress-responsive OsvWA genes had either very few TE insertions mostly primarily in the non-coding regions or very low insertion allele frequency. In contrast, non-expressing genes harbored transposons in the CDS region. This indicated that either the non- expressing genes have accumulated more TEs or the insertion of more elements has rendered them non-functional. For example, in this study, all the genes showing significant up-regulation in response to blast infection either had TE insertions in introns (,and) or had no TE insertions (,and). Further, the expression ofand, the only genes with two TE insertions in CDS region, was not detected in the qRT-PCR analysis after blast infection in rice. Moreover, their expression was not detected in any available transcriptome databases. Thus, these two genes could be non-expressing vWA members in rice.
Besides the TEs, SNPs determine the expression as well as the function of the genes. vWA genes in Tetep were highly diverse from Nipponbare and HP2216 and showed a large number of SNPs. Tetep is resistant to various biotic stresses and shows significant differences in gene sequence compared to HP2216 and Nipponbare (Balint-Kurti, 2019; Wang et al, 2019; Kumar et al, 2021). Additionally, vWA genes in both HP2216 and Nipponbare are highly similar and barely have any SNPs, explaining their similar phenotypes in stress response (Chen et al, 2019). The SNPs identified in this study along with the distribution of these genes on different chromosomes would help to identify/develop markers that can also be utilized in marker-assisted breeding programs.
vWA genes in rice had other domains in addition to vWA domain. The most important one is the RING finger domain, present in as many as 16 genes which is known to be actively involved in mediating protein-protein interactions and functioning in E3 ubiquitin-protein ligase activity (Freemont, 2000). OsvWA9 contains three UIM domains and codes for 26S proteasome non-ATPase regulatory subunit 4 and thus, it has a role in protein degradation pathways. The UIM is found in ubiquitin-associated proteins and is responsible for ubiquitin recognition (Polo et al, 2002). This ubiquitin recognition function coincides with the MapMan analysis where maximum genes were found to be involved in the proteolysis pathway. The ubiquitin recognition and proteolysis function predicted that these genes may interact with several other proteins and could play a significant role in signal transduction or degradation of proteins through proteasome-mediated protein degradation pathway. OsvWA5 (OsBON1) and OsvWA16 (OsBON3) proteins, which are homologs of AtBON1 and AtBON3, respectively, have two C2 domains and function as a negative regulator of bacterial and fungal disease resistance (Yin et al, 2018). vWA domain of AtBON1 protein participates in protein-protein interactions to recruit other proteins at the cell membrane. qRT-PCR expression analysis ofandgenes in this study showed slight up-regulation in blast infected tissues in both Tetep and HP2216 varieties. Earlier reports showed semi-dwarf and lower tillering phenotype of transgenic plants overexpressinggene (Yin et al, 2018). Therefore, plants express the gene at a low level to maintain the balance between disease response with normal growth and proper tillering. Genevestigator data showed slight up-regulation and no change in expression ofininoculated varieties Pusa Basmati and near isogenic line (NIL) Pusa Basmati containinggene, respectively. Thus, all these results showed no drastic up- or down-regulation of the gene under biotic stress, indicating genotype specific expression.() is expressed in response to both biotic and abiotic stimuli and therefore, it is an important gene to explore modified plant’s response to various stresses (Kazan, 2017).
Expression ofgene in callus and cell culture and also in response to cytokinin in roots indicates its role in cell division and totipotency(Hirose et al, 2007).andshow higher expression in sheath, panicle and shoot. The presence of these genes in blast-resistantlocus also explains their expression in panicle and shoot (Fang et al, 2019).andgenes also show enhanced expression in response to. qRT-PCR analysis of blast infected panicle tissues in this study showed induced expression of these genes. These two genes were also highly up-regulated in transcriptome data of rice panicles after infection of(Kumar et al, 2021). In addition to blast response,andgenes are expressed in response to gall midge insect, a parasitic weed, and also after infection ofpv.. All these results make these two genes potential biotic stress-responsive genes that can be exploited for utilization in breeding programs.was also up-regulated during salt stress at the vegetative stage, depicting its role in abiotic stress.
Genevestigator analysis indicated induced expression ofgene after infection ofandpv.. In addition, its high expression in rice panicles afterinfection confirms its role in biotic stress response. Expression ofgene increased in Tetep as well as HP2216 after blast disease infection where its expression was higher in Tetep compared to HP2216. OsvWA10 protein is localized in the plasma membrane and predicts to have protein binding activity. It is annotated to play a role in the protein modification process. The expression data on the Rice Genome Annotation Project reveals that it is only expressed in panicles, shoots and pre-emergence inflorescences. Thus, panicle blast responsive expression of this gene observed in this study suggests its role in panicle blast resistance.
Considering the expression of vWA genes during theinfection of important rice diseases, we have eventually shortlisted the genes that may play a significant role in biotic stress response.,andgenes were expressed specifically in response to blast and bacterial blight whereaswas common in bacterial blight and sheath blight, showing a complex pattern of expression against biotic stress. However,andshowed induced expression in all these three diseases, making them important genes for response to biotic stress, and it needs further studies for understanding their exact roles and mechanisms of disease response.
METHODS
Identification and structural analysis of vWA domain- containing genes in rice
InterProScan tool (v5.44-79.0) (https://www.ebi.ac.uk/interpro/ search/sequence/) was used for identifying the domains of amino acid sequences. Different domain databases such as CDD (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml), Pfam (http://pfam.xfam.org/) and SMART (http://smart.embl- heidelberg.de/) were also considered as references. The vWA domains were selected based on the keyword (vWA) and accession IDs (PF00092, SM000327 and IPR002035). Output was generated in a TSV (Tab Separated Value) format. The resulting file was loaded into Microsoft Excel for further analysis.
Protein sequences of the vWA domain-containing genes in rice were retrieved from Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/). The protein sequences were then analyzed for identification and structural analysis of motifs present in the proteins in addition to vWA domain using the SMART tool. In addition, the transposon insertion polymorphism in all the OsvWA genes was analyzed using the RTRIP database (Liu et al, 2020). SNPs in the genomic sequence of all these genes in two rice varieties Tetep and HP2216, in comparison to Nipponbare sequence, were also analyzed manually by aligning the sequences using BioEdit software. Subcellular localization of all the vWA proteins was predicted by the WoLF PSORT Protein Subcellular Localization Prediction Tool (Horton et al, 2007).
Chromosomal distribution of OsvWA genes
The physical position of all vWA domain-containing genes were obtained from the Rice Genome Annotation Project. The chromosomal location image of these genes was created using the Mapchart 2.32 software (Voorrips, 2002).
Phylogenetic analysis and evolutionary relationship
Multiple sequence alignment of protein sequences of all the vWA domain-containing genes was done using the MUSCLE tool in MEGA7 (Edgar, 2004). The phylogenetic tree was constructed based on the alignment file using the neighbor joining method with 1000 bootstrap replications in MEGA7 (Kumar et al, 2016). Sequence similarity among the OsvWA members was identified by generating Circos plot (Krzywinski et al, 2009). In addition to evolutionary relationships among protein sequences, the promoter sequences of all the genes in Tetep and Nipponbare varieties were also studied by phylogenetic analysis. For this purpose, 1 kb promoter sequence of the genes was retrieved from the genome sequences and the phylogenetic tree was generated by following the same procedure as described for the protein sequences.
Expression analysis of OsvWA genes during environmental stress and plant development
Expression of all the OsvWA genes at different developmental stages, different tissues, and during different environmental stress responses was analyzed with Genevestigator (Hruz et al, 2008) by selecting development, anatomy and perturbations respectively, in the search tool. Both microarray and RNA-Seq databases were explored for expression analysis. Besides Genevestigator, expression analysis of these genes was carried out by exploring the RiceMetaSys database for salt and drought stress (Sandhu et al, 2017) and RiceMetaSysB database for biotic stress namely blast and bacterial blight (Sureshkumar et al, 2019). Additionally, the involvement of OsvWA genes and their expression in various metabolic pathways were investigated by using the MapMan tool (Thimm et al, 2004). Gene expression values from previous transcriptome data of blast infected tissues (48 hpi) of rice were used for the analysis.
Plant materials and disease infection
Two rice varieties, Tetep and HP2216, were grown in a glasshouse under controlled conditions with (25 ± 2) ºC and 16 h day/ 8 h night cycle till the reproductive stage. For preparing the fungal inoculums, Mo-ni-025, a most virulent strain ofwas cultured on potato dextrose agar at 25 ºC for 10–12 d. The fungus was then transferred to Mathur’s media for induction of reproductive growth. After 8–10 d of reproductive growth, it was scrapped by adding 5 mL of autoclaved ddH2O and used for the preparation of a conidial suspension of approximately 1 × 105cfu/mL. Rice panicles were inoculated with conidial suspension using the syringe inoculation method by injecting them at the neck of the panicles. Plants were kept under controlled conditions with higher humidity for enhanced pathogen infection. Rice panicles were collected at 48 hpi along with mock and immediately dipped in liquid nitrogen and stored at -80 ºC.
RNA isolation, cDNA synthesis, and qRT-PCR analysis
Total RNA from all the collected panicle samples was isolated using the Spectrum Plant Total RNA Kit (Sigma-Aldrich, St. Louis, Missouri, USA) according to the manufacturer’s protocol. The quality and quantity of the isolated RNA were assessed by gel electrophoresis and using a Nano-Drop spectrophotometer 2000 (Thermo Fisher Scientific, Wilmington, USA). cDNA synthesis was carried out from 1 µg of total RNA using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, California, USA) according to the manufacturer’s protocol. qRT-PCR was carried out to analyze the expression of the vWA genes in the samplesgene was used as an internal control. Primers for qRT-PCR assay were designed using the PrimerQuest tool (https://eu.idtdna.com/) and all the primer sequences were shown in Table S5. Brilliant III Ultra-Fast Sybr®Green QPCR Master Mix (Agilent Technologies, USA) was used for qRT-PCR and the reaction was carried out in Light-Cycler®480 II (Roche, Rotkreuz, Switzerland). Each 10 µL qRT-PCR reaction mix contained 0.1 µL of each primer, 5 µL of 2× SYBR Green Master Mix, 0.15 µL of ROX fluorescence dye, 1 µL of diluted cDNA, and 3.65 µL of nuclease-free water. All the reactions were carried out using three biological replicates with three technical replicates. The thermal cycler program used for the reaction was 95 ºC for 30 s, 60 ºC for 15 s, and 72 ºC for 20 s with 40 cycles of amplification. The single melt cycle from 65 ºC to 95 ºC was used at the end to get the dissociation curve. To analyze the differential expression of genes in samples, 2–ΔΔCT(Cycle threshold) method was followed (Livak and Schmittgen, 2001).
ACKNOWLEDGEMENTS
The study was supported by the Indian Council of Agricultural Research (ICAR)-National Institute for Plant Biotechnology, National Agricultural Higher Education Project: Centre for Advanced Agricultural Science and Technology (Grant No. 1010033), and ICAR-Centre for Agricultural Bioinformatics, Indian Agricultural Statistics Research Institute, New Delhi(IASRI) (Grant No. 1006456).
SUPPLEMENTAL DATA
The following materials are available in the online version of this article at http://www.sciencedirect.com/journal/rice-science; http://www.ricescience.org.
Fig. S1. Domains in vWA proteins predicted by conserved domain database search using SMART tool.
Fig. S2. Circos plot showing duplication events during the evolution of vWA gene family.
Fig. S3. Expression of OsvWA genes at different rice developmental stages.
Fig. S4. Expression of OsvWA genes in different rice tissues.
Fig. S5. Expression of OsvWA genes in rice tissues under different treatments resulting in biotic or abiotic stress.
Fig. S6. Mapman pathway analysis showing the role of vWA proteins in various cellular pathways along with their expression levels.
Table S1. Structural details and subcellular localization of OsvWA genes.
Table S2. Transposon insertion polymorphism in OsvWA genes.
Table S3. Single nucleotide polymorphism details of vWA genes with reference to Nipponbare sequence.
Table S4. Gene expression data of vWA genes observed in RiceMetaSys and RiceMetaSysB databases.
Table S5. List of primers used for qRT-PCR analysis.
Balint-Kurti P. 2019. The plant hypersensitive response: Concepts, control and consequences., 20(8): 1163‒1178.
Bharati K P, Prashanth U R. 2011. Von Willebrand disease: An overview., 73(1): 7‒16.
Chen X L, Jia Y L, Wu B M. 2019. Evaluation of rice responses to the blast fungusat different growth stages., 103(1): 132‒136.
Edgar R C. 2004. MUSCLE: Multiple sequence alignment with high accuracy and high throughput., 32(5): 1792‒1797.
Fang N Y, Wei X R, Shen L T, Yu Y, Li M Y, Yin C F, He W W, Guan C H, Chen H, Zhang H S, Bao Y M. 2019. Fine mapping of a panicle blast resistance geneinlandrace Bodao and its application in rice breeding., 12(1): 18.
Freemont P S. 2000. Ubiquitination: RING for destruction?, 10(2): R84‒R87.
Hirose N, Makita N, Kojima M, Kamada-Nobusada T, Sakakibara H. 2007. Overexpression of a type-A response regulator alters rice morphology and cytokinin metabolism., 48(3): 523‒539.
Horton P, Park K J, Obayashi T, Fujita N, Harada H, Adams-Collier C J, Nakai K. 2007. WoLF PSORT: Protein localization predictor., 35: W585‒W587.
Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W, Zimmermann P. 2008. Genevestigator v3: A reference expression database for the meta-analysis of transcriptomes., 2008: 420747.
Kazan K. 2017. The multitalented MEDIATOR25., 8: 999.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S J, Marra M A. 2009. Circos: An information aesthetic for comparative genomics., 19(9): 1639‒1645.
Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets., 33(7): 1870‒1874.
Kumar V, Jain P, Venkadesan S, Karkute S G, Bhati J, Abdin M Z, Sevanthi A M, Mishra D C, Chaturvedi K K, Rai A, Sharma T R, Solanke A U. 2021. Understanding rice:interaction in resistant and susceptible cultivars of rice under panicle blast infection using a time-course transcriptome analysis., 12(2): 301.
Li W T, Chern M, Yin J J, Wang J, Chen X W. 2019. Recent advances in broad-spectrum resistance to the rice blast disease., 50: 114‒120.
Liu J X, Jambunathan N, McNellis T W. 2005. Transgenic expression of the von Willebrand A domain of the BONZAI 1/ COPINE 1 protein triggers a lesion-mimic phenotype in., 221(1): 85‒94.
Liu Z, Wang T Z, Wang L, Zhao H, Yue E K, Yan Y, Irshad F, Zhou L, Duan M H, Xu J H. 2020. RTRIP: A comprehensive profile of transposon insertion polymorphisms in rice., 18(12): 2379‒2381.
Livak K J, Schmittgen T D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCTmethod., 25(4): 402‒408.
Polo S, Sigismund S, Faretta M, Guidi M, Capua M R, Bossi G, Chen H, de Camilli P, Di Fiore P P. 2002. A single motif responsible for ubiquitin recognition and monoubiquitination in endocytic proteins., 416: 451‒455.
Rawat N, Naga N C, Meenakshi S R, Nair S, Bentur J S. 2012. A novel mechanism of gall midge resistance in the rice variety Kavya revealed by microarray analysis., 12(2): 249‒264.
Reininger A J. 2008. Function of von Willebrand factor in haemostasis and thrombosis., 14: 11‒26.
Sandhu M, Sureshkumar V, Prakash C, Dixit R, Solanke A U, Sharma T R, Mohapatra T, Sv A M. 2017. RiceMetaSys for salt and drought stress responsive genes in rice: A web interface for crop improvement., 18(1): 432.
Sureshkumar V, Dutta B, Kumar V, Prakash G, Mishra D C, Chaturvedi K K, Rai A, Sevanthi A M, Solanke A U. 2019. RiceMetaSysB: A database of blast and bacterial blight responsive genes in rice and its utilization in identifying key blast-resistant WRKY genes., 2019: baz015.
Swarbrick P J, Huang K, Liu G, Slate J, Press M C, Scholes J D. 2008. Global patterns of gene expression in rice cultivars undergoing a susceptible or resistant interaction with the parasitic plant., 179(2): 515‒529.
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller L A, Rhee S Y, Stitt M. 2004. MAPMAN: A user‐driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes., 37(6): 914‒939.
Tuckwell D. 1999. Evolution of von Willebrand factor A (VWA) domains., 27(6): 835‒840.
Voorrips R E. 2002. MapChart: Software for the graphical presentation of linkage maps and QTLs., 93(1): 77‒78.
Wang L, Zhao L N, Zhang X H, Zhang Q J, Jia Y X, Wang G, Li S M, Tian D C, Li W H, Yang S H. 2019. Large-scale identification and functional analysis ofgenes in blast resistance in the Tetep rice genome sequence., 116(37): 18479‒18487.
Whittaker C A, Hynes R O. 2002. Distribution and evolution of von Willebrand/integrin A domains: Widely dispersed domains with roles in cell adhesion and elsewhere., 13(10): 3369‒3387.
Yang S H, Yang H J, Grisafi P, Sanchatjate S, Fink G R, Sun Q, Hua J. 2006. Thegene family represses cell death and promotes cell growth in., 45(2): 166‒179.
Yin X, Zou B H, Hong X X, Gao M J, Yang W B, Zhong X B, He Y, Kuai P, Lou Y G, Huang J R, Hua J, He Z H. 2018. Rice copine genesandfunction as suppressors of broad-spectrum disease resistance., 16(8): 1476‒1487.
Yu J, Wang J, Lin W, Li S G, Li H, Zhou J, Ni P X, Dong W, Hu S N, Zeng C Q, Zhang J G, Zhang Y, Li R Q, Xu Z Y, Li S T, Li X R, Zheng H K, Cong L J, Lin L, Yin J N, Geng J N, Li G Y, Shi J P, Liu J, Lv H, Li J, Wang J, Deng Y J, Ran L H, Shi X L, Wang X Y, Wu Q F, Li C F, Ren X Y, Wang J Q, Wang X L, Li D W, Liu D Y, Zhang X W, Ji Z D, Zhao W M, Sun Y Q, Zhang Z P, Bao J Y, Han Y J, Dong L L, Ji J, Chen P, Wu S M, Liu J S, Xiao Y, Bu D B, Tan J L, Yang L, Ye C, Zhang J F, Xu J Y, Zhou Y, Yu Y P, Zhang B, Zhuang S L, Wei H B, Liu B, Lei M, Yu H, Li Y Z, Xu H, Wei S L, He X M, Fang L J, Zhang Z J, Zhang Y Z, Huang X G, Su Z X, Tong W, Li J H, Tong Z Z, Li S L, Ye J, Wang L S, Fang L, Lei T T, Chen C, Chen H, Xu Z, Li H H, Huang H Y, Zhang F, Xu H Y, Li N, Zhao C F, Li S T, Dong L J, Huang Y Q, Li L, Xi Y, Qi Q H, Li W J, Zhang B, Hu W, Zhang Y L, Tian X J, Jiao Y Z, Liang X H, Jin J, Gao L, Zheng W M, Hao B L, Liu S Q, Wang W, Yuan L P, Cao M L, McDermott J, Samudrala R, Wang J, Wong G K S, Yang H M. 2005. The genomes of: A history of duplications., 3(2): e38.
11 August 2021;
12 November 2021
Amolkumar U. Solanke (amol.solanke@icar.gov.in)
Copyright © 2022, China National Rice Research Institute. Hosting by Elsevier B V
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
Peer review under responsibility of China National Rice Research Institute
http://dx.doi.org/10.1016/j.rsci.2021.11.007
(Managing Editor: Wang Caihong)

- Rice Science的其它文章
- RPA-Assisted Cas12a System for Detecting Pathogenic Xanthomonas oryzae, a Causative Agent for Bacterial Leaf Blight Disease in Rice
- Polycomb Repressive Complex 2-Mediated H3K27 Trimethylation Is Required for Pathogenicity in Magnaporthe oryzae
- Selenium Alleviates Carbohydrate Metabolism and Nutrient Composition in Arsenic Stressed Rice Plants
- Feasibility of Improving Unmanned Aerial Vehicle-Based Seeding Efficiency by Using Rice Varieties with Low Seed Weight
- Genetic Variation for Anaerobic Germination and Emergence from Deeper Soil Depth in Oryza nivara Accessions
- Arsenic Accumulation in Rice:Sources, Human Health Impact and Probable Mitigation Approaches