
Polycomb Repressive Complex 2-Mediated H3K27 Trimethylation Is Required for Pathogenicity in Magnaporthe oryzae
2022-06-16 11:37WuZhonglingQiuJiehuaShiHuanbinLinChuyuYueJiangnanLiuZhiquanXieWeiNaweedNaqviKouYanjunTaoZeng
Rice Science 2022年4期
Wu Zhongling, Qiu Jiehua, Shi Huanbin, Lin Chuyu, Yue Jiangnan, Liu Zhiquan, Xie Wei, Naweed I. Naqvi, Kou Yanjun, Tao Zeng
Research Paper
Polycomb Repressive Complex 2-Mediated H3K27 Trimethylation Is Required for Pathogenicity in
Wu Zhongling1, #, Qiu Jiehua2, #, Shi Huanbin2, Lin Chuyu1, Yue Jiangnan1, Liu Zhiquan2, Xie Wei1, Naweed I. Naqvi3, Kou Yanjun2, Tao Zeng1
(Ministry of Agriculture and Rural Affairs Key Laboratory of Molecular Biology of Crop Pathogens and Insects, Institute of Biotechnology, Zhejiang University, Hangzhou 310058, China; State Key Laboratory of Rice Biology, China National Rice Research Institute, Hangzhou 311400, China; Temasek Life Sciences Laboratory and Department of Biological Sciences, 1 Research Link, National University of Singapore, Singapore 117604, Singapore; These authors contributed equally to this work)
Polycomb repressive complex 2 (PRC2) contributes to catalyze the methylation of histone H3 at lysine 27 and plays vital roles in transcriptional silencing and growth development in various organisms. In, histone H3K27 is found to associate with altered transcription ofinduced genes. However, it is still unknown whether and how H3K27me3 modification is involved in pathogenicity to rice and stress response. In this study, we found that core subunits of PRC2, Kmt6-Suz12-Eed, were required for fungal pathogenicity to rice in. Kmt6-Suz12-Eed localized in the nuclei and was necessary for the establishment of H3K27me3 modification. With ChIP-seq analysis, 9.0% of genome regions enriched with H3K27me3 occupancy, which corresponded to 1033 genes in. Furthermore, deletion of,oraltered genome-wide transcriptional expression, while the de-repression genes in the ∆strain were highly associated with H3K27me3 occupancy. Notably, plenty of genes which encode effectors and secreted enzymes, secondary metabolite synthesis genes, and cell wall stress-responsive genes were directly occupied with H3K27me3 modification and de-repression inthe ∆strain. These results elaborately explained how PRC2 was required for pathogenicity, which is closely related to effector modulated host immunity and host environment adaption.
rice blast; H3K27me3; transcriptional regulation; pathogenicity; Polycomb repressive complex 2
Organisms need to reprogram gene expression properly in different development stages or environment stimuli, which suppressing unnecessary genes is the most important process (Netea et al, 2016; Cavalli and Heard, 2019). Generally, stable maintenance of repressed gene expression states is achieved partially by the propagation of specific chromatin modifications (Cavalli and Heard, 2019). Polycomb repressive complex (PRC) plays a vital role in such repression and contributes to facultative heterochromatin in the chromosome (Margueron and Reinberg, 2011; Schuettengruber et al, 2017; Wiles and Selker, 2017). PRC is involved in various biological processes, including maintaining cellular and tissue identity in multicellular organisms and regulating phase transitions in plants (Margueron and Reinberg, 2011; Blackledge et al, 2015). In mammals and higher plants, there are two main Polycomb group complexes, PRC1 and PRC2 (Lanzuolo and Orlando, 2012). PRC1, including Pc, Ph and Psc, compacts chromatin and catalyzes the monoubiquitylation of histone H2A. PRC2, including core subunit proteins, Ezh (Kmt6), Su(z)12, Esc (Eed) and additional subunits RbAp48/Nurf55 (P55), catalyzes the methylation of histone H3 at lysine 27 (Schuettengruber et al, 2017; Wiles and Selker, 2017). The core subunits of PRC2 are conserved fromto mammals and higher plants, while the PRC1 components are not evolutionarily conserved. Notably, PRC1 has not been identified in fungi so far (Margueron and Reinberg, 2011; Ridenour et al, 2020).
In the fungal kingdom, disruption of PRC usually reprograms genome-wide transcriptional expression, leading to abnormal growth and reduced pathogenicity (Wiles and Selker, 2017; Ridenour et al, 2020). In wheat head blight pathogen, deletion of PRC2 core subunits results in complete loss of H3K27me3 modification, up-regulation of about 2 500 genes, as well as severe defects in fungal growth and pathogenicity (Connolly et al, 2013). In, loss of PRC2core subunits also abolishes all H3K27me3 modifications, but only accompanied by slight growth defects (Jamieson et al, 2013). In symbiotic fungus, H3K27me3 coupled with H3K9me3 controls the expression of symbiosis-specific genes, such as genes related to alkaloid bioprotective metabolites (Chujo and Scott, 2014). In, deletion of,or additional subunit, removes all H3K27me3 modifications (Dumesic et al, 2015). In the soybean root rot pathogen, occupancy of H3K27me3 at the effector geneleads to transcriptional silencing and fails to induce themediated disease resistance (Wang et al, 2020), which highlights that epigenetic variation is an effective adaptive strategy in plant-pathogen interaction. All these studies suggest that PRC2 has diverse functions in different fungi, and its biological functions in pathogenic fungi still need to be further studied (Wiles and Selker, 2017; Ridenour et al, 2020).
Rice blast, caused by, is a devastating rice disease and threatens food security worldwide. During the infection of, the three-celled conidia germinate and form appressoria to penetrate the rigid rice cuticle under proper conditions. Once inside the host cell,differentiates into invasive hyphae and spreads to neighboring cells, thus forming rice blast lesion. During invasive growth,needs to coordinate the gene expression regulation, stress response, and eluding the plant immunity to adapt to the host milieu. As we know that deletion ofinresults in a decrease in sporulation and highly reduced pathogenicity in barley (Pham et al, 2015). However, the molecular mechanisms involved in regulating of PRC2 mediated H3K27me3 modification during invasive growth has not been explored in depth. Recently, Zhang et al (2021) elucidated that histone dynamics of H3K27 is associated with altered transcription ofinduced genes, including effectors,in. Whether H3K27me3 modification is involved in other biological processes such as pathogenicity to rice and stress response are still unknown.
RESULTS
PRC2 is required for pathogenicity in M. oryzae
Polycomb-group genes are first identified inand are usually assembled with Polycomb PRC1 and PRC2 (Blackledge et al, 2015; Ridenour et al, 2020). To identify the candidate subunits of PRC1 and PRC2 in, BLASTp was performed to search for the orthologs from70-15 genomes (taxid: 242507) with the query sequences from,and(Table S1). Three unique PRC2 core subunits, Kmt6 (MGG_00152), Eed (MGG_06028) and Suz12 (MGG_03169), were obtained, while no PRC1 subunit was hit even by low stringency BLAST search in(Table S1). To analyze the function of PRC2, deletion mutants of,andwere created by the homologous recombination strategy instrain B157 (Fig. S1-A and -B). As reported, the PRC2 mutants showed defects in the mycelia growth and conidiation formation(Fig. S2-A to -C) (Zhang et al, 2021), suggesting that the events of gene replacement were correct. Moreover, the complementary strains Δ-C,Δ-C and Δ-C were obtained by introducing the wild type (WT),andloci into their corresponding mutants. The resultant complementarystrains were similar with WT strain in colony morphology, indicating that Δ-C, Δ-Cand Δ-C functionally complemented the phenotypes of Δ, Δand Δ.
To further analyze the function of PRC2 subunits in pathogenicity to rice, the conidia of WT, Δ, Δ, Δand their complementary strains were collected and inoculated on the seedlings of the susceptible rice cultivar CO39. As the number of conidia from the mutants was highly reduced (15%‒ 25% of WT) (Fig. 1-A), low concentration of conidia (5 × 104cfu/mL) was used in the rice seedling infection assay. Compared with WT that caused the characteristic spindle-shaped blast lesions with grey centres, the Δ, Δand Δstrains formed relative fewer and restricted lesions (Fig. 1-B). To further decipher these observations, the appressorium formation on the inductive hydrophobic surface and rice sheath was investigated. Although no obvious change in appressorium formation on the rice sheath was found in the mutantstrains, the invasive growth displayed significant difference between the mutants and WT (Fig. 1-C). Nearly 90% appressorium from the WT strain successfully penetrated the rice sheath, while only 60% appressorium from the deletion mutants were capable of penetration (Fig. 1-C). Moreover, the formation of secondary invasive hypha in the mutant strains was less than that of WT. These results suggested that PRC2 is necessary for penetration and invasive hyphal growth.

Fig. 1. Polycomb repressive complex 2 (PRC2) is required for fungal pathogenicity in.
A, Relative lesion area of indicated strains. The relative lesion area was quantified by an ImageJ software. Δ, Δand Δare deletion mutants from the wild type (WT) strain. Δ-C, Δ-C and Δ-C are their complementary strains, respectively.
B, Blast infection assay using 21-day-old rice seedlings (CO39). Deletion of,andimpaired the pathogenicity of. Scale bars, 2 mm.
C,Observation and statistical analysis of invasive hypha growth in rice sheath cells at 40 h post-inoculation. Four types of invasive hyphae: no penetration (Type I), penetration with primary hyphae (Type II), with differentiated secondary invasive hyphae (Type III), and invasive hyphae spreading into neighbouring cells (Type IV), were quantified. Data represent Mean ± SD of three independent repeats, with= 300 appressoria per analysis. Scale bars, 5 μm.
PRC2 core subunits are indispensable for H3K27me3 activity in M. oryzae
To elucidate the function of PRC2 core subunits during pathogenesis in, we first fused Kmt6, Suz12 or Eed with green fluorescent protein (GFP) and examined their subcellular localization in. As shown in Fig. 2-A, all these subunits were co-localized with Hoechst-stained nuclei (Fig. 2-A). Then, yeast two-hybrid assay was conducted to test whether these subunits are physically associated. The results showed that at least Kmt6 can interact with Eed in the yeast cells, suggesting that these components likely form a complex in the nuclei as PRC2 in other species (Fig. 2-B). Subsequently, the protein levels of histone lysine methylation were detected with specific antibodies using the Western blotting assay. The levels of H3K27me3 were almost undetectable in the ∆, ∆and ∆mutants, while the decreased levels of H3K27me3 were completely restored in the complementary strains respectively (Fig. 2-C). Meanwhile, the levels of H3K4me3 and H3K36me3 had no obvious changes in all the aforementioned strains compared with WT (Fig. 2-D). We concluded that Suz12 and Eed, as well as Kmt6, are fully and specifically required for PRC2-mediated H3K27me3 modification in.
Core subunits of PRC2 regulate genome-wide gene expression in M. oryzae
As H3K27me3 modification often associates with transcriptional silencing on target genes, we explored the roles of PRC2 in the transcriptional regulation inusing high-throughout sequencing (RNA- seq) at the vegetative mycelia with three biological repeats. As expected, totally 19.2%, 18.6% and 20.1% of genome-wide genes were identified as up-regulated genes with equal or greater than 2-fold change (log2> 1,< 0.05), while only 5.3%, 6.0% and 8.8% of genes were identified as down-regulated genes in the ∆,∆and ∆strains, respectively (log2< -1,< 0.05) (Fig. 3-A). Furthermore, ∆,∆and∆shared 80%‒86% overlap (2 144 genes) in their up- regulated genes (Fig. 3-B). These results further confirmed that core subunits of PRC2 regulate similar biological processes and H3K27me3 modification plays conserved roles as transcriptional repressors in.
To explore the detailed biological process involved in PRC2, Gene Ontology (GO) analysis was conducted with up-regulated genes in the,andstrains. Biological process as carbohydrate metabolic process, transmembrane transport, interaction with host via secreted proteins and metabolic process were significantly enriched in the three PRC2 deletion mutants (Fig. 3-C). These results implied that PRC2- mediated H3K27me3 modification is involved in extensive biological processes including pathogenicity in.
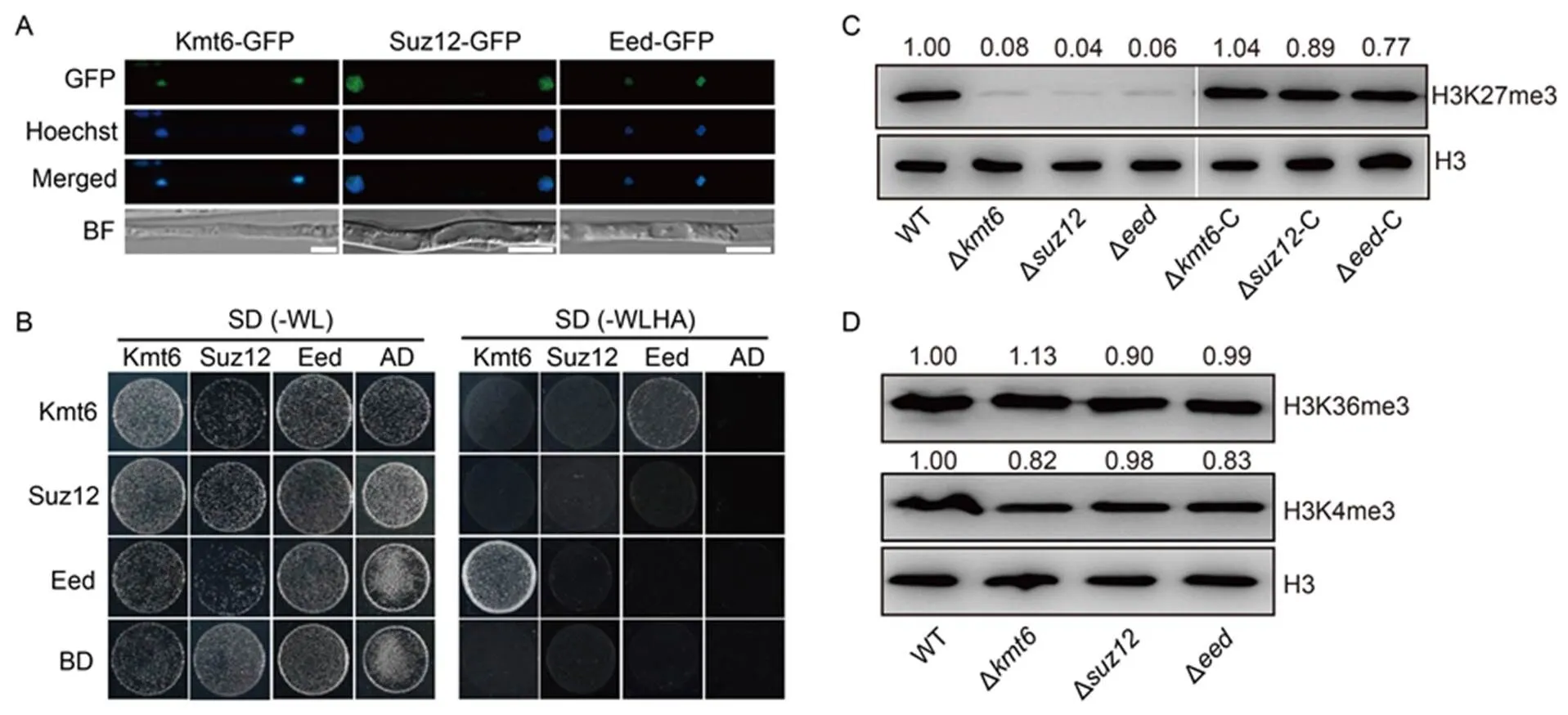
Fig. 2. Core subunits of Polycomb repressive complex 2 (PRC2) are indispensable for H3K27me3 modification in.
A, Confocal microscopy image for subcellular localization of PRC2 subunits fused with green fluorescent protein (GFP). The GFP signal co-localized with Hoechst (10 μg/mL) stained nuclei. BF, Blight field. Scale bars, 5 μm.
B, Yeast two-hybrid assay of PRC2 components Kmt6, Eed and Suz12. The bait and prey plasmids were co-transformed into yeast strain Y2Hgold, respectively. Then, the transformants were grown on basal medium SD (-WL, without tryptophan and leucine) and selective medium SD (-WLHA, without tryptophan, leucine, histidine and adenine). The empty plasmids pGADT7 (AD) and pGBKT7 (BD) were used as controls.
C, Levels of histone H3 and H3K27me3 in wild type (WT), ∆, ∆and∆strains and their complementary strains (∆-C, ∆-C and∆-C) on total histones were measured by Western blotting. The relative intensity abundance was measured and calculated. Two repeated biological experiments were conducted with similar results.
D, Levels of histone H3, H3K4me3 and H3K36me3 in WT, ∆, ∆and∆strains were measured by Western blotting. The relative intensity abundances were measured and calculated. Two repeated biological experiments were conducted with similar results.
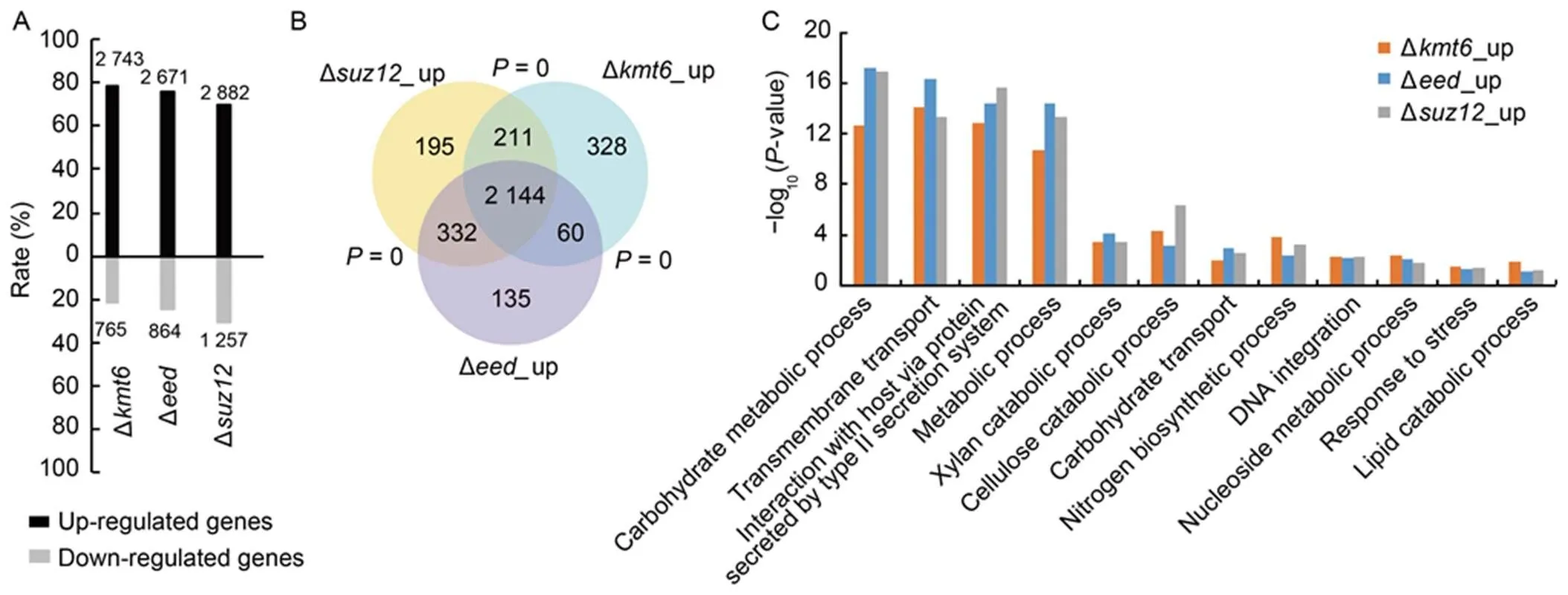
Fig. 3. Core subunits of Polycomb repressive complex 2 (PRC2) regulate genome-wide gene expression in.
A, Summary of up- and down-regulated genes in different PRC2 deletion mutants with RNA-seq analysis. The numbers at the top or bottom of the bars are the numbers of differential expressed genes in the mutants. The-axis is the percentage of up- and down-regulated genes in all the differentially expressed genes. Data were obtained from three independent biological repeats.
B, Venn diagram showing statistically significant overlaps among gene sets of ∆_up,∆_upand∆_up.value for overlapping between gene sets was obtained by the Fisher’s exact test.
C, Gene Ontology (GO) analysis of ∆_up,∆_upand∆_up genes.
H3K27me3 occupancy is highly associate with de-repression of target genes in PRC2 deletion mutants
To further investigate how H3K27me3 associated with transcriptional regulation in, we mapped the genome-wide H3K27me3 occupancy using the chromatinimmunoprecipitation assay followed by high-throughout sequencing (ChIP-seq) in the WT and ∆strains. The intensity of H3K27me3 occupancy in the ∆mutant was barely detectable (Fig. 4-A), which was consistent with the role of Kmt6 as H3K27me3 writer inUsing the ∆as background, 9.0% of genome regions enriched with H3K27me3 occupancy in the WT strain (Fig. S3-A). Totally, 1 082 significant peaks were identified in the WT strain (log2> 1,< 0.05), which mainly spread within gene bodies and were corresponded to 1 033 genes (Fig. 4-B).
To verify whether up-regulated genes in ∆(∆up) were directly associated with the loss of H3K27me3 occupancy, the gene sets between ∆up and H3K27me3-marked genes were compared. With low threshold (log2> 1,< 0.05), 23.5% differential genes in ∆were marked with H3K27me3 occupancy(Fig. 4-B). Meanwhile, with stringent threshold (log2> 3,< 0.05), 40.1% differential genes in ∆were marked with H3K27me3 (Fig. 4-B). Meanwhile, 22.2% (641 of 2 882) and 23.3% (625 of 2 671) gens in ∆_upand∆up respectively were remarkably associated with H3K27me3 occupancy (Fig. S3-B). Together, the de-repressed genes in the mutants of PRC2 core subunits are highly associated with the absence of H3K27me3 occupancy in.
Next, GO analysis was conducted with H3K27me3- occupancy genes. Biological process such as metabolic process, transmembrane transport and interaction with host via secreted proteins were significantly enriched (Fig. S3-C), which was similar with the results from ∆up. To validate the ChIP-seq results, two effector genes (and) and two cutinase genes (and) were presentedfor further analysis (Mosquera et al, 2009; Sharpee et al, 2017). The chromatin of these genes enriched with H3K27me3 occupancy in WT but nearly undetectable in the ∆mutant (Fig. 4-C). Independent ChIP- qPCR experiments were also convinced that the de-repression of examined genes in thestrain accompanied with almost undetectable H3K27me3 occupancy (Fig. 4-D). These results suggested that H3K27me3 modification is tightly associated with transcriptional repression of target genes through direct occupancy on the chromatin.
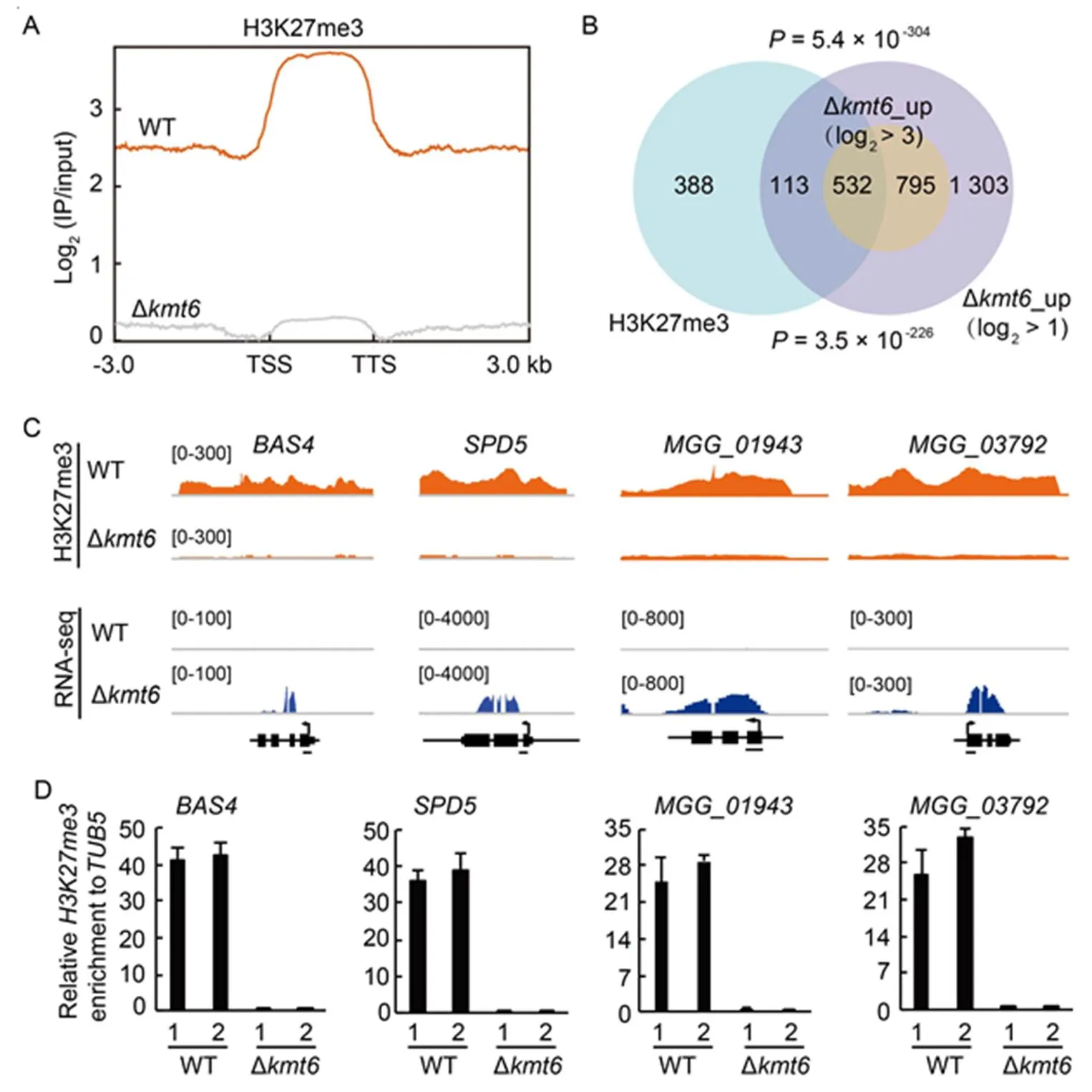
Fig. 4. H3K27me3 occupancy is highly associated with de-repression of target genes in Polycomb repressive complex 2 (PRC2) deletion mutants.
A,Average H3K27me3 occupancy within 3-kb genomic regions flanking the summit of H3K27me3 peaks in wild type (WT) and ∆strains. TSS, Transcription start site; TTS, Transcription termination site.
B, Venn diagram showing statistically significant overlaps between gene sets of H3K27me3-occupied genes and_up with thresholds of log2> 1 and log2> 3 respectively.values with Fisher’s exact test for overlapping between gene sets were labeled.
C, Genome browser views of H3K27me3 occupancy from chromatin immune- precipitation assay followed by high- throughout sequencing (ChIP-seq) and expression pattern from RNA-seq of selected genes. The number areas were reads per million (RPM).
D, ChIP-qPCR verified the enrichment of H3K27me3 at the chromatin of selected genes. The examined regions were shown with black line at the bottom of gene model. The relative enrichments were calculated by relative quantitation from two biological repeats, which was standardized with an internal control, then compared with that of WT. The numbers ‘1’ and ‘2’ indicated two independent repeats. Values areMean ± SD of three technical repeats.
Expression change on plenty of virulence genes may contribute to PRC2 associated-pathogenicity
Combined with RNA-seq and ChIP-seq analyses, we found that plenty of genes which encode effectors and other virulence factors were directly occupied with H3K27me3 modification and de-repressed inthe∆strain compared with WT. To verify these results, representative types of genes were further investigated with independent experiments in the ∆and WT strains.
During plant-pathogen interaction, pathogens secrete effectors to modulate plant immunity and ensure progressive infection (Fouché et al, 2018). Two() genes,and, which encode effectors during the vegetative growth stage (Mosquera et al, 2009; Khang et al, 2010; Sharpee et al, 2017), exhibited significantly increased expression levels in ∆than those in WT (Fig. 5-A). To further explore whether PRC2 regulates genome-wide effector expression, gene sets of putative genome-wide effectors, including 247 effectors (only 246 had reads in the RNA-seq experiments) were extracted for further analysis (Dong et al, 2015; Sharpee et al, 2017). Consistent with recent findings (Zhang et al, 2021), 51.2% (126/246) effectors exhibited significantly increased expression (log2> 1,< 0.05) in the ∆strain (Fig. S4). To further explore whether the increased transcriptional expression of effectorswould enhance accumulation and secretion of effectors during the vegetative growth stage, BAS4-GFP construct was introduced into the WT and ∆strains. As shown in Fig. 5-B, Bas4-GFP signal was evident in the conidia and the apical and subapical regions of the hyphae in the ∆strain, but can not be detectable in those of WT (Fig. 5-B). These findings indicated that the loss of H3K27me3 occupancy reprograms the expression of large number of effectors and results in untimely expression of effectors during the vegetative growth stage, which was similar with the natural expression at the invasive growth stage (Fig. 5-B).
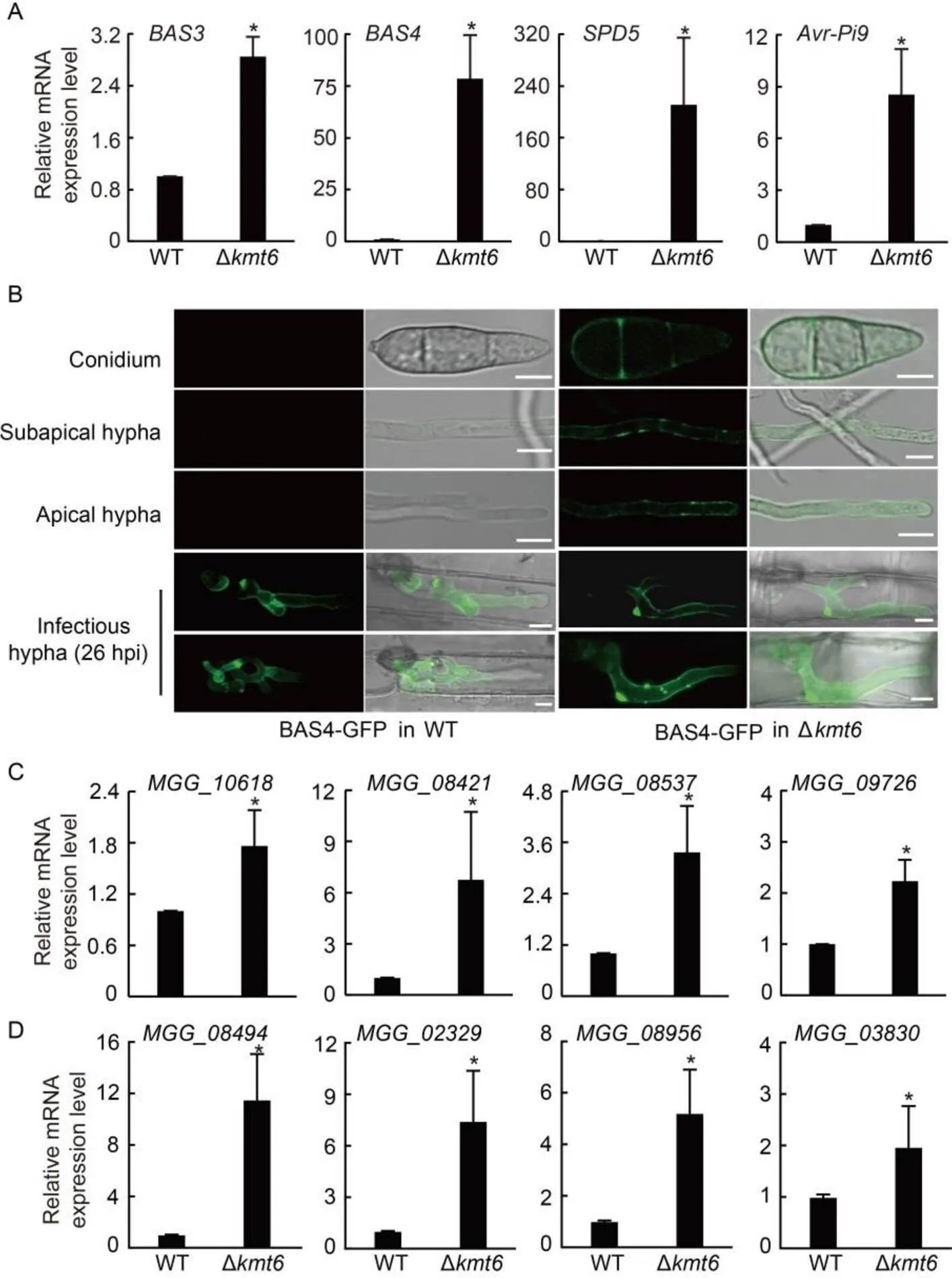
Fig. 5. Expression levels of genes which encode effector, extracellular enzyme and cytochrome P450 were up-regulated in Polycomb repressive complex 2 (PRC2) deletion mutants.
A,Relative expression levels of effectors were checked in wild type (WT) and Δstrains. The strains were cultured in liquid complete medium (CM) at 28 ºC for 2 d.was used as an internal reference.
B, Confocal microscopy image of Bas4- GFP at the vegetative andgrowth stages of WT and ∆strains. In WT, Bas4-GFP fluorescence was only shown at thegrowth stage, while in ∆, Bas4-GFP fluorescence can be detected in the conidia, apical hyphae, subapical hyphaeand invasive hyphae. GFP, Green fluorescent protein; hpi, Hours post-inoculation. Scale bars, 5 μm.
C and D,Relative expression levels of pathogenic genes that encode extracellular enzyme (C) and cytochrome P450 (D) were checked in WT and Δstrains.was used as an internal reference. The strains were cultured in liquid CM at 28 ºC for 2 d.
Values areMean ± SD of three biological repeats. *, Significant differences at< 0.05 between the deletion mutant and WT strains by the Student’s-test.
Secreted enzymes and cytochrome P450 genes are involved in secondary metabolite synthesis and virulence in pathogenic fungi (Oh et al, 2008; Kawahara et al, 2012). In addition to effectors, four secreted enzyme genes (,_,and_) and four P450 genes including,_,and_were checked by the qRT-PCR assay. These genes exhibited 5‒40-fold increased expression levels in ∆than in WT (Fig. 5-C and -D), suggesting that secreted enzymes and some secondary metabolite synthesis genes may also contribute to PRC2 associated- pathogenicity to rice.
PRC2 is required for cell wall stress response in M. oryzae
Pathogen needs to endure various abiotic stresses from the host and environment to successfully survive and accomplish pathogenic infection.has evolved and acquired the ability to response proper cell wall stress (Feng et al, 2021; Qian et al, 2021). In addition to effectors and some virulence genes, we noted that plenty of cell wall stress-responsive genes were directly occupied with H3K27me3 modification and de-repression in the∆strain compared with WT. To verify these results, the expression of four cell-wall responsive genes including_,_,_and_(Oh et al, 2008) were also confirmed to be up-regulated in the ∆mutant (Fig. 6-A). To further investigate whether PRC2 core subunits associated with cell wall stress response, the sensitivities of ∆, ∆, ∆and their complementary strains on the complete medium supplemented with 0.005% sodium dodecyl sulphate (SDS), 0.1 mg/mL Congo red (CR), or 0.05 mg/mL calcofluor white (CFW) were tested. The results showed that the growth of deletion mutants ∆, ∆and ∆exhibited increased sensitivity to CFW and CR conditions compared to the WT strain(Fig. 6-B to -E), indicating that PRC2 is required for cell wall stress response in.
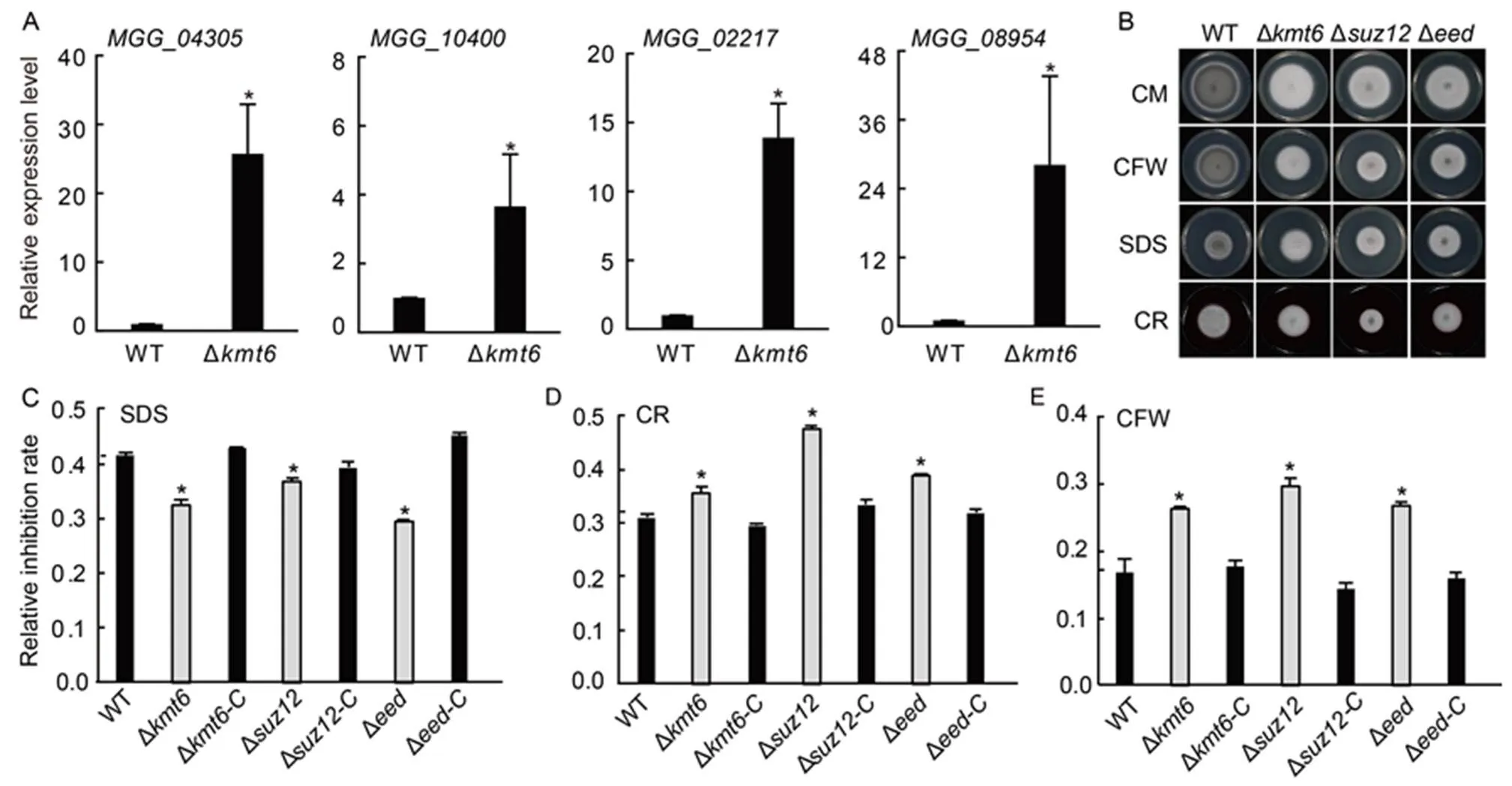
Fig. 6. Polycomb repressive complex 2 (PRC2) is required for stress response in.
A,Relative expression levels of cell wall related genes. Values areMean ± SD of three biological repeats. *, Significant differences at< 0.05 between the deletion mutant and wild type (WT) strains by the Student’s-test. The strains were cultured in liquid CM at 28 ºC for 2 d.was used as an internal reference.
B,Radical growths of WT, ∆, ∆, ∆and their complementary strains (∆-C, ∆-C and∆-C). Colonies of indicated strains were grown on CM supplemented with different chemicals for 7 d.
C‒E, Statistical analysis of colony diameters of tested strains on CM supplemented with 0.005% SDS (C), 0.1 mg/mL CR (D), and 0.05 mg/mL CFW (E) for 7 d.Values areMean ± SD of three independent repeats. *, Significant differences at< 0.05 between the deletion mutant and WT strains by the Student’s-test.
CM, Complete medium; SDS, Sodium dodecyl sulphate; CR, Congo red; CFW, Calcofluor white.
DISCUSSION
H3K27me3 modification is well characterized to associate with facultative heterochromatin and genome stability, subsequently plays vital roles in transcriptionalregulation and development of the fungi (Ridenour et al, 2020). Although Zhang et al (2021) reported histone H3K27 inis associated with altered transcription ofinduced genes, whether H3K27me3 modification involves in other biological processes such as pathogenicity to rice and stress responses are still unknown. In this study, we found that PRC2 core subunits Kmt6-Suz12-Eed mediated H3K27me3 modification, and were required for pathogenicity to rice in. Furthermore, Kmt6-Suz12-Eed regulated genome-wide transcriptional silencing and H3K27me3 occupancy were highly associated with de-repression of targeted genes in the PRC2 deletion mutants. These target genes included virulence genes and stress responsive genes, which likely contributed to PRC2 associated-pathogenicity to rice.
Epigenetic modification is correlated with genome plasticity and stability, and helps pathogen effective adaption to host environments (Chadha and Sharma, 2014; Cavalli and Heard, 2019; Wang et al, 2020). In, epigenetic modifications like histone (de)methylation and (de)acetylation have extensively been explored in different biological processes such as infection structure morphogenesis, development transition and autophagy. Histone deacetylase genesandregulate infectious growth and asexual development in(Ding et al, 2010; Lee et al, 2019). H3K4 methyltransferase Set1 contributes to appressorium formation, conidiation and pathogenicity with transcriptional activation on infection-related genes (Pham et al, 2015). Histone acetyltransferase Gcn5 negatively regulates light-induced autophagy and conidiation through acetylating the autophagy protein Atg7 (Zhang et al, 2017).plays a key role in infection-associated autophagy and infection through recruiting histone deacetylase complex on the target chromatin (He et al, 2018). H3K27me3 modification and transcriptional silencing, which are mediated by PRC2 subunits Kmt6-Suz12-Eed, have been extensively studied in multicellular organisms (Margueron and Reinberg, 2011; Schuettengruber et al, 2017; Wiles and Selker, 2017; Ridenour et al, 2020). In, we found that each of the core subunits Kmt6-Suz12-Eed had a unique homologue and was required for H3K27me3 modification. Furthermore, deletion mutants exhibited impaired mycelia growth, conidiation, pathogenicity, as well as response to cell wall stress (Figs. 1, 6 and S1), implying that PRC2 regulates the expression of plenty of genesBased on the RNA-seq and ChIP-seq analyses, we concluded thatKmt6-Suz12-Eed functions as a conserved transcriptional repressor which directly associates with H3K27me3 occupancy to conduct Polycomb silencing on the targeted genes. However, how these core subunits work together and how to establish and maintain H3K27me3 modification in the fungal are still largely unknown.
Establishment and maintenance H3K27me3 to local chromatin accompany with reprogramming of gene expression, depends on histone ‘reader’ and a series of/-regulatory factors which act in corporate or alone (Kassis and Brown, 2013; Xiao et al, 2017). Recently, a conserved bromo-adjacent homology (BAH) domain and a plant homeodomain (PHD) finger protein were identified from plants and fungi with ability to recognize H3K27me3 and mediate genome- wide Polycomb silencing (Li et al, 2018; Yang et al, 2018; Wiles et al, 2020). As the destined gene, the mechanism is more complicated and coordinates with various factors according to local chromatin background.
Pathogen effectors are rapidly evolved, especially distribute in regions with high genome plasticity. In, effectorsdo not have common motifs or conserved-elements responsible for their adaptabilities to host environment (Dong et al, 2016; Sánchez-Vallet et al, 2018). This study and previous analysis appeared to support that H3K27me3 preferentially modifies the poorly conserved or newly evolved effectors to ‘guard’genome and save energy through transcriptional silencing (Dong et al, 2015; Fouché et al, 2018; Sánchez-Vallet et al, 2018). In addition, secreted enzymes, secondary metabolite synthesis genes and cell wall stress responsive genes are also regulated by H3K27me3 modification (Figs. 5 and 6). It seemed that whenever necessary, such as in response to the host environment, fungal pathogens would redistribute H3K27me3 modification and evoke transcriptional activation machinery, then activate effectors and other virulence genes to help pathogen better and effectively adapt and colonize in the host (Galazka and Freitag, 2014; Fouché et al, 2018). These H3K27me3 modifications regulate the expression of effectors, secreted enzymes, secondary metabolite synthesis genes and cell wall stress response genes, elaborately explain how PRC2 is required for pathogenicity, which is closely related to effector modulated host immunity and host environment adaption.
In conclusion, PRC2 core subunits Kmt6-Suz12- Eed mediated H3K27me3 modification is required for pathogenicity to rice likely through regulating the expression of effectors, some virulence genes and cell wall stress responsive genes in.
METHODS
Fungal strains and culture conditions
WT strain B157 was used as a background strain for obtaining transformants, which was a kind gift from the Indian Institute of Rice Research (Hyderabad, India). For growth assessment of, strains were grown on the complete medium (CM) for 7 d (Kou et al, 2017). For stress assessment, strains were grown on CM supplemented with 0.005% SDS, 0.1 mg/mL CR, or 0.05 mg/mL CFW at 25ºC in the dark for 7 d. For conidiation,strains were grown on CM at 25 ºC in the dark for 2 d, followed by growth under continuous lights for 5 d.
Plasmid construction
To create the deletion mutants of,and, the standard one-step gene replacement strategy was used. Briefly, approximate 1-kb of 5′ UTR and 3′ UTR regions were amplified and ligated sequentially to the flanking of,orgene cassette in the pFGL821 (58223, Addgene, USA), pFGL820 (58222, Addgene, USA) or pFGL822(58225, Addgene, USA), respectively (Kou et al, 2017). The sequences of plasmids were confirmed by sequencing and subsequently introduced into the B157 strain bymediated transformation (MT). To generate the localization and complementary vectors, theandterminators were cloned to pFGL820 to obtainterminator. The fragments containing about 1.5-kb of promoter and coding region were amplified and cloned into theterminator. After confirmed by sequencing, the resultant plasmids were introduced into deletion mutants respectively byMT. Primers used in this study were listed in Table S2 and strains were listed in Table S3
Live cell imaging and image processing
The CM cultivated hyphae were stained with 10 μg/mL Hoechst 33342 (Sigma, USA) for 20 min to visualize nuclei. Live cell epifluorescence microscopy was performed with LSM700 (CarlZeiss Inc, Germany), using the requisite conditions established for detecting GFP or Hoechst signals. Image processing was performed using ImageJ (http://fiji.sc/wiki/ index.php/Fiji).
Rice seedling and rice sheath infection assay
Rice seedling infection assays were performed with 5 × 104cfu/mL conidial suspension as described by Kou et al (2017). Disease symptoms of infection assays were assessed on 7 d post-inoculation. The infection assays were repeated three times. For theinvasive hyphal development assay, 21-day- old healthy rice seedlings (CO39) were used for sheath preparation. Conidial suspension (5 × 104cfu/mL) was inoculated into the rice sheath and incubated in the growth chamber with the photoperiod of 16-h light and 8-h dark at 25 ºC. The inoculated sheath was trimmed manually and observed using an Olympus BX53 wide field microscope equipped with bright field optics (OLYMPUS, Beijing, China) at 40 h post-inoculation.
Yeast two-hybrid analysis
Yeast two-hybrid assays were performed in the yeast strain Y2Hgold with the Matchmaker two-hybrid system (Clontech, USA) according to the manufacturer’s instruction. The baits and preys were cloned to pGADT7 and pGBKT7, respectively. The transformants were grown on the basic medium without tryptophan and leucine at 30 ºC for about 2 d, and subsequently the direct interaction between two proteins were tested with grown on selective medium without tryptophan, leucine, histidine and adenine for 4‒6 d.
Western blotting
For detecting histones, 0.5 g mycelia cultured in the liquid CM for 2 d were collected. The nuclei of mycelia were isolated with extraction buffer (20 mmol/L Tris, pH 7.5, 20 mmol/L KCl, 2 mmol/L MgCl2, 25% glycerol, 250 mmol/L sucrose, 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF), 5 mmol/L beta-mercaptoethanol, and 1× proteinase inhibitor) and filtered through two layers of Miracloth (Millipore, USA). Then, the histones were extracted with lysis buffer (50 mmol/L Tris-HCl, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, 1% Triton X-100, and 1× protein inhibitor). Total histones were separated by 15% SDS-PAGE gel and protein blots were detected with anti-H3 (06-755, Millipore, USA), anti-H3K27me3 (ab6002, Abcam, UK), anti-H3K4me3 (ab1012, Abcam, UK) and anti-H3K36me3 (ab9050, Abcam, UK). The relative intensity of Western blots was quantified by the ImageJ software.
mRNA expression analysis with qRT-PCR
Total RNA was extracted from CM cultivated mycelia for 2 d from three biological repeats using TRIzol reagent (Invitrogen, USA). Subsequently, total RNA was reversely transcribed into cDNAs with commercial kits (FSQ-301, TOYOBO, Japan) according to the manufacturer’s instruction. qRT-PCR was performed using SYBR Green qPCR Master Mix (QST-100, TOYOBO, Japan) in the LightCycler480 system (Roche, USA). The constitutively expressedgene () was used as endogenous control to normalize the amount of cDNA templates. Primers used were listed and described in Table S2.
RNA-seq analysis
Total RNA was extracted from mycelia from three biological repeats using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s instruction. RNA was sequenced separately at Novogene, using Illumina Hiseq X-Ten with the Hiseq-PE150 strategy. The clean reads were mapped to the reference genome of(70-15 assembly MG8 from NCBI) using the TopHat software with default settings (Kim et al, 2013). For analysis of differentially expressed genes, the assembled transcripts from three independent biological replicates in the WT and deletion mutants were included and compared using Cuffdiff with default settings (Trapnell et al, 2010). Gene with false discovery rate threshold of 0.05, in conjunction with at least 2-fold change in expression level was considered differentially expressed. To calculate the significance of the overlap of two gene sets,value with Fisher’s exact test for overlapping was performed online (the total number of genes in thegenome used was 14 317) (http:// nemates.org/MA/progs/overlap_stats.html). GO analysis for enriched biological processes was performed using DAVID (https://david.ncifcrf.gov/home.jsp) with default settings.
ChIP and ChIP-seq analysis
TheChIP experiments with mycelia were conducted as previous reports with minor modification (Tao et al, 2017; He et al, 2018). Briefly, 1 g mycelia were crosslinked with 1% formaldehyde for 20 min and stopped with 125 mmol/L glycine for 5 min at room temperature. Samples were ground with liquid nitrogen and resuspended in the nucleus isolating buffer (10 mmol/L Tris, pH 8.0, 10 mmol/Lsodium butyrate, 400 mmol/Lsucrose, 0.1 mmol/L PMSF, 5 mmol/Lbeta- mercaptoethanol, and 1× proteinase inhibitor). Subsequently, the precipitated nuclei were used to total chromatin extraction with 1 mL lysis buffer (50 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 10 mmol/Lsodium butyrate, 0.1% deoxycholate, 0.1% SDS, 1% Triton X-100, 1 mmol/L PMSF, and 1× Roche protease inhibitor cocktail). The lysis chromatin was sonicated into DNA fragments between 200‒ 500 bp using Diagenode Bioruptor (high setting, 16 cycles, every cycle with 30 s ‘on’ and 30 s ‘off’). About 20 μL chromatin was used to input DNA extraction and the remainder was pre-cleared with 10 μL protein A Dynabeads (10001D, Thermofisher, USA) for 1 h. Then, the chromatin was incubated with anti-H3K27me3 (ab6002, Abcam, UK) overnight at 4 ºC. Another 20 μL protein A Dynabeads was used to capture protein-DNA mixture and followed by three washing. Protein-DNA mixture was reverse-crosslinked, and DNA was recovered with phenol-chloroform extraction. The recovered DNA was used as a template for followed ChIP- qPCR and ChIP-seq. Two biological repeats were conducted.
For ChIP-seq assay, the purified DNA was used as library construction with the NEB Next Ultra II DNA Library Prep Kit for Illumina (E7645L, NEB, USA). High-throughput sequencing was carried out using Illumina Hiseq-PE150 by Novogene Corporation (Beijing, China) for Illumina (Langmead et al, 2009). Subsequently, the clean read pairs were mapped to the reference genome with Bowtie2 (Version 2.2.8) (Langmead and Salzberg, 2012) and enriched peaks were called by MACS2 (Version 2.1.1) with default parameters (Zhang et al, 2008). The data were imported into the integrative genomics viewer for visualization (Robinson et al, 2011). To assign peaks to proximal genes, the gneomic sequence of 3-kb flanking the peak summit was extracted. To validate ChIP-seq results, ChIP-qPCR assay with two independent repeats was performed. The levels of examined fragments were relative to an internal reference geneusing qRT-PCR. The PCR primers were listed and described in Table S2.
Data availability
The ChIP-seq and RNA-seq datasets generated were deposited in the Gene Expression Omnibus (GEO) under the accession number GSE166690.
ACKNOWLEDGEMENTS
This study was supported in part by the National Natural Science Foundation of China (Grant Nos. 32170192 and 32000103), Zhejiang Science and Technology Major Program on Agricultural New Variety Breeding (Grant No. 2021C02064), Key Research and Development Project of China National Rice Research Institute (Grant No. CNRRI-2020-04), the Chinese Academy of Agricultural Sciences under the ‘Elite Youth’ Program and the Agricultural Sciences and Technologies Innovation Program. Thanks to Dr. Zhou Ming of Zhejiang University for help of bioinformatics analysis, Dr. Qi Zhenyu of agricultural experimental station of Zhejiang University for controlling the light environment of artificial climate chamber.
SUPPLEMENTAL DATA
The following materials are available in the online version of this article at http://www.sciencedirect.com/journal/rice-science; http://www.ricescience.org.
Fig. S1. Identification of Polycomb repressive complex 2 deletion mutants.
Fig. S2. Polycomb repressive complex 2 is required for fungal growth and conidium formation in.
Fig. S3. ChIP-seq analysis of wild type and Polycomb repressive complex two deletion mutants.
Fig. S4. Distribution of 246 effectors based on expression change in ∆compared with their wild type using RNA- seq analysis.
Table S1. Components of Polycomb repressive complex in,,and.
Table S2. Primers used in this study.
Table S3. Strains used in this study.
Blackledge N P, Rose N R, Klose R J. 2015. Targeting Polycomb systems to regulate gene expression: Modifications to a complex story., 16: 643‒649.
Cavalli G, Heard E. 2019. Advances in epigenetics link genetics to the environment and disease., 571: 489‒499.
Chadha S, Sharma M. 2014. Transposable elements as stress adaptive capacitors induce genomic instability in fungal pathogen., 9: e94415.
Chujo T, Scott B. 2014. Histone H3K9 and H3K27 methylation regulates fungal alkaloid biosynthesis in a fungal endophyte- plant symbiosis., 92: 413‒434.
Connolly L R, Smith K M, Freitag M. 2013. Thehistone H3 K27 methyltransferase KMT6 regulatesdevelopment and expression of secondary metabolite gene clusters., 9: e1003916.
Ding S L, Liu W D, Iliuk A, Ribot C, Vallet J, Tao A, Wang Y, Lebrun M H, Xu J R. 2010. The Tig1 histone deacetylase complex regulates infectious growth in the rice blast fungus., 22: 2495‒2508.
Dong Y H, Li Y, Zhao M M, Jing M F, Liu X Y, Liu M X, Guo X X, Zhang X, Chen Y, Liu Y F, Liu Y H, Ye W W, Zhang H F, Wang Y C, Zheng X B, Wang P, Zhang Z G. 2015. Global genome and transcriptome analyses ofepidemic isolate 98-06 uncover novel effectors and pathogenicity- related genes, revealing gene gain and lose dynamics in genome evolution., 11: e1004801.
Dong Y H, Li Y, Qi Z Q, Zheng X B, Zhang Z G. 2016. Genome plasticity in filamentous plant pathogens contributes to the emergence of novel effectors and their cellular processes in the host., 62: 47‒51.
Dumesic P A, Homer C M, Moresco J J, Pack L R, Shanle E K, Coyle S M, Strahl B D, Fujimori D G, Yates III J R, Madhani H D. 2015. Product binding enforces the genomic specificity of a yeast Polycomb repressive complex., 160: 204‒218.
Feng W Z, Yin Z Y, Wu H W, Liu P, Liu X Y, Liu M X, Yu R, Gao C Y, Zhang H F, Zheng X B, Wang P, Zhang Z G. 2021. Balancing of the mitotic exit network and cell wall integrity signaling governs the development and pathogenicity in., 17: e1009080.
Fouché S, Plissonneau C, Croll D. 2018. The birth and death of effectors in rapidly evolving filamentous pathogen genomes., 46: 34‒42.
Galazka J M, Freitag M. 2014. Variability of chromosome structure in pathogenic fungi: Of ‘ends and odds’., 20: 19‒26.
He M, Xu Y P, Chen J H, Luo Y, Lv Y, Su J, Kershaw M J, Li W T, Wang J, Yin J J, Zhu X B, Liu X H, Chern M, Ma B T, Wang J C, Qin P, Chen W L, Wang Y P, Wang W M, Ren Z L, Wu X J, Li P, Li S G, Peng Y L, Lin F C, Talbot N J, Chen X W. 2018. MoSnt2-dependent deacetylation of histone H3 mediates MoTor- dependent autophagy and plant infection by the rice blast fungus., 14: 1543‒1561.
Jamieson K, Rountree M R, Lewis Z A, Stajich J E, Selker E U. 2013. Regional control of histone H3 lysine 27 methylation in., 110: 6027‒6032.
Kassis J A, Brown J L. 2013. Polycomb group response elements inand vertebrates., 81: 83‒118.
Kawahara Y, Oono Y, Kanamori H, Matsumoto T, Itoh T, Minami E. 2012. Simultaneous RNA-seq analysis of a mixed transcriptome of rice and blast fungus interaction., 7: e49423.
Khang C H, Berruyer R, Giraldo M C, Kankanala P, Park S Y, Czymmek K, Kang S, Valent B. 2010. Translocation ofeffectors into rice cells and their subsequent cell-to-cell movement., 22: 1388‒1403.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg S L. 2013. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions., 14: R36.
Kou Y J, Tan Y H, Ramanujam R, Naqvi N I. 2017. Structure- function analyses of the Pth11 receptor reveal an important role for CFEM motif and redox regulation in rice blast., 214: 330‒342.
Langmead B, Salzberg S L. 2012. Fast gapped-read alignment with Bowtie 2., 9: 357‒359.
Langmead B, Trapnell C, Pop M, Salzberg S L. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome., 10: R25.
Lanzuolo C, Orlando V. 2012. Memories from the polycomb group proteins., 46: 561‒589.
Lee J, Lee J J, Jeon J. 2019. A histone deacetylase, MoHOS2 regulates asexual development and virulence in the rice blast fungus., 57: 1115‒1125.
Li Z C, Fu X, Wang Y Z, Liu R Y, He Y H. 2018. Polycomb- mediated gene silencing by the BAH-EMF1 complex in plants., 50: 1254‒1261.
Margueron R, Reinberg D. 2011. The Polycomb complex PRC2 and its mark in life., 469: 343‒349.
Mathioni S M, Patel N, Riddick B, Sweigard J A, Czymmek K J, Caplan J L, Kunjeti S G, Kunjeti S, Raman V, Hillman B I, Kobayashi D Y, Donofrio N M. 2013. Transcriptomics of the rice blast fungusin response to the bacterial antagonistreveals candidate fungal defense response genes., 8: e76487.
Mosquera G, Giraldo M C, Khang C H, Coughlan S, Valent B. 2009. Interaction transcriptome analysis identifiesBAS1-4 as biotrophy-associated secreted proteins in rice blast disease., 21: 1273‒1290.
Netea M G, Joosten L A B, Latz E, Mills K H G, Natoli G, Stunnenberg H G, O'Neill L A, Xavier R J. 2016. Trained immunity: A program of innate immune memory in health and disease., 352: aaf1098.
Oh Y, Donofrio N, Pan H Q, Coughlan S, Brown D E, Meng S W, Mitchell T, Dean R A. 2008. Transcriptome analysis reveals new insight into appressorium formation and function in the rice blast fungus., 9: R85.
Pham K T M, Inoue Y, Vu B V, Nguyen H H, Nakayashiki T, Ikeda K I, Nakayashiki H. 2015. MoSET1 (histone H3K4 methyltransferase in) regulates global gene expression during infection-related morphogenesis., 11: e1005385.
Qian B, Liu X Y, Ye Z Y, Zhou Q K, Liu P, Yin Z Y, Wang W H, Zheng X B, Zhang H F, Zhang Z G. 2021. Phosphatase- associated protein MoTip41 interacts with the phosphatase MoPpe1 to mediate crosstalk between TOR and cell wall integrity signalling during infection by the rice blast fungus., 23: 791‒809.
Ridenour J B, Möller M, Freitag M. 2020. Polycomb repression without bristles: Facultative heterochromatin and genome stability in fungi., 11(6): 638.
Robinson J T, Thorvaldsdóttir H, Winckler W, Guttman M, Lander E S, Getz G, Mesirov J P. 2011. Integrative genomics viewer., 29: 24‒26.
Sánchez-Vallet A, Fouché S, Fudal I, Hartmann F E, Soyer J L, Tellier A, Croll D. 2018. The genome biology of effector gene evolution in filamentous plant pathogens., 56: 21‒40.
Schuettengruber B, Bourbon H M, di Croce L, Cavalli G. 2017. Genome regulation by polycomb and trithorax: 70 years and counting., 171: 34‒57.
Sharpee W, Oh Y, Yi M, Franck W, Eyre A, Okagaki L H, Valent B, Dean R A. 2017. Identification and characterization of suppressors of plant cell death (SPD) effectors from., 18: 850‒863.
Tao Z, Shen L S, Gu X F, Wang Y Z, Yu H, He Y H. 2017. Embryonic epigenetic reprogramming by a pioneer transcription factor in plants., 551: 124‒128.
Trapnell C, Williams B A, Pertea G, Mortazavi A, Kwan G, van Baren M J, Salzberg S L, Wold B J, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation., 28: 511‒515.
Villalba F, Collemare J, Landraud P, Lambou K, Brozek V, Cirer B, Morin D, Bruel C, Beffa R, Lebrun M H. 2008. Improved gene targeting inby inactivation of MgKU80 required for non-homologous end joining., 45: 68‒75.
Wang L Y, Chen H, Li J J, Shu H D, Zhang X X, Wang Y C, Tyler B M, Dong S M. 2020. Effector gene silencing mediated by histone methylation underpins host adaptation in an oomycete plant pathogen., 48: 1790‒1799.
Wiles E T, Selker E U. 2017. H3K27 methylation: A promiscuous repressive chromatin mark., 43: 31‒37.
Wiles E T, McNaught K J, Kaur G, Selker J M L, Ormsby T, Aravind L, Selker E U. 2020. Evolutionarily ancient BAH-PHD protein mediates Polycomb silencing., 117: 11614‒11623.
Xiao J, Jin R, Yu X, Shen M, Wagner J D, Pai A, Song C, Zhuang M, Klasfeld S, He C S, Santos A M, Helliwell C, Pruneda-Paz J L, Kay S A, Lin X W, Cui S J, Garcia M F, Clarenz O, Goodrich J, Zhang X Y, Austin R S, Bonasio R, Wagner D. 2017.anddeterminants of epigenetic silencing by Polycomb repressive complex 2 in., 49: 1546‒1552.
Yang Z L, Qian S M, Scheid R N, Lu L, Chen X S, Liu R, Du X, Lv X C, Boersma M D, Scalf M, Smith L M, Denu J M, Du J M, Zhong X H. 2018. EBS is a bivalent histone reader that regulates floral phase transition in., 50: 1247‒1253.
Zhang S L, Liang M L, Naqvi N I, Lin C X, Qian W Q, Zhang L H, Deng Y Z. 2017. Phototrophy and starvation-based induction of autophagy upon removal of Gcn5-catalyzed acetylation of Atg7 in., 13: 1318‒1330.
Zhang W, Huang J, Cook D E. 2021. Histone modification dynamics at H3K27 are associated with altered transcription ofinduced genes in., 17: e1009376.
Zhang Y, Liu T, Meyer C A, Eeckhoute J, Johnson D S, Bernstein B E, Nusbaum C, Myers R M, Brown M, Li W, Liu X S. 2008. Model-based analysis of ChIP-Seq (MACS)., 9: R137.
20 August 2021;
23 November 2021
Tao Zeng (taozeng@zju.edu.cn); Kou Yanjun(kouyanjun@caas.cn)
Copyright © 2022, China National Rice Research Institute. Hosting by Elsevier B V
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
Peer review under responsibility of China National Rice Research Institute
http://dx.doi.org/
(Managing Editor: Wu Yawen)

- Rice Science的其它文章
- RPA-Assisted Cas12a System for Detecting Pathogenic Xanthomonas oryzae, a Causative Agent for Bacterial Leaf Blight Disease in Rice
- Genome-Wide Analysis of von Willebrand Factor A Gene Family in Rice for Its Role in Imparting Biotic Stress Resistance with Emphasis on Rice Blast Disease
- Selenium Alleviates Carbohydrate Metabolism and Nutrient Composition in Arsenic Stressed Rice Plants
- Feasibility of Improving Unmanned Aerial Vehicle-Based Seeding Efficiency by Using Rice Varieties with Low Seed Weight
- Genetic Variation for Anaerobic Germination and Emergence from Deeper Soil Depth in Oryza nivara Accessions
- Arsenic Accumulation in Rice:Sources, Human Health Impact and Probable Mitigation Approaches