
RPA-Assisted Cas12a System for Detecting Pathogenic Xanthomonas oryzae, a Causative Agent for Bacterial Leaf Blight Disease in Rice
2022-06-16 11:35KittisakBuddhachatNattapornSripairojOnchiraRitbamrungPhithakInthimaKumropRatanasutThanitaBoonsrangsomTepsudaRungratPongsanatPongcharoenKaweeSujipuli
Rice Science 2022年4期
Kittisak Buddhachat, Nattaporn Sripairoj, Onchira Ritbamrung, Phithak Inthima, Kumrop Ratanasut, Thanita Boonsrangsom, Tepsuda Rungrat, Pongsanat Pongcharoen, Kawee Sujipuli
Research Paper
RPA-Assisted Cas12a System for Detecting Pathogenic, a Causative Agent for Bacterial Leaf Blight Disease in Rice
Kittisak Buddhachat1, Nattaporn Sripairoj1, Onchira Ritbamrung1, Phithak Inthima1, Kumrop Ratanasut2, 3, Thanita Boonsrangsom2, 3, Tepsuda Rungrat2, 3, Pongsanat Pongcharoen2, 3, Kawee Sujipuli2, 3
(Department of Biology, Faculty of Science, Naresuan University, Phitsanulok 65000, Thailand; Center of Excellence in Research for Agricultural Biotechnology, Department of Agricultural Science, Faculty of Agriculture, Natural Resources and Environment, Naresuan University, Phitsanulok 65000, Thailand; Department of Agricultural Science, Faculty of Agriculture, Natural Resources and Environment, Naresuan University, Phitsanulok 65000, Thailand)
pv.() is a widespread pathogen causing bacterial leaf blight (BLB) disease, devastating rice productivity in many cultivated areas of Thailand. A specific and simple method fordetection is required to improve surveillance of disease transmission and outbreak. This study developed arecombinase polymerase amplification (RPA) assay assisted with CRISPR-cas12a assay (RAC) fordetection from bacterial cell suspension of infected rice samples without DNA extraction.The efficiency of the RAC system fordetection using eitherorlocus was optimized to amplify and determine the sensitivity and specificity using aDNA template from bacterial cell suspension of infected rice samples without DNA extraction. The RAC system using thelocus gave a higher specificity thanlocus, because onlyspecies was amplified positive RPA product with fluorescence signal by cas12a digestion, which indicated no cross reactivity. Optimal RAC using thelocus enabled diagnosis ofpresence from both plant extracted samples ofartificially inoculated rice leaves within 3 d post-inoculationwithout symptomatic BLB appearance, andnaturally infected rice. Findings exhibited that RAC using thelocus offered sensitivity, specificity and simplicity fordetection, with low intensities of-DNA (1 × 103copies/µL) and-cell (2.5 × 103cfu/mL). This developed RAC system showed significantly potential fordetection at point-of-care application for early signs of BLB disease outbreak in rice fields.
bacterial leaf blight; CRISPR-cas12a; recombinase polymerase amplification; rice
pv.() is a disease- causing agent of bacterial leaf blight (BLB) symptoms in rice, resulting in severe crop yield losses in Asia, including Thailand (Yasmin et al, 2017). However, effective strategies have not yet been developed to control bacterialepidemics in rice fields (le Thanh et al, 2017). To avert outbreak and transmission ofin rice for long-term control of BLB disease, the development of a specific, sensitive and rapid nucleic acid approach is imperative for earlydetection before visual appearance of BLB symptoms.
PCR assays including conventional PCR (cPCR) (Lang et al, 2010), multiplex PCR (mPCR) (Cui et al, 2016) and real-time quantitative PCR (qRT-PCR) (Lu et al, 2014) have been developed as reliable diagnostic methods fordetection, corresponding to highly specific regions ofandloci (Lang et al, 2010). These assays have been widely used to detect several DNA microorganisms, however, cPCR, mPCR and qRT-PCR detection assays require specially equipped laboratories (with thermocyclers and real- time PCR machines) and special technical expertise (Garita-Cambronero et al, 2017) that impede their practical use in rice fields. By contrast, new isothermalamplification technology allows nucleic acid amplificationunder a constant temperature has been gaining attentionfor pathogen detection such as loop mediated isothermal amplification (LAMP) (Notomi et al, 2000; Buddhachatet al, 2021) and recombinase polymerase amplification (RPA) (Piepenburg et al, 2006; Daher et al, 2016). This technique has provided potential use for on-site diagnosis compared to cPCR, mPCR and qRT-PCR technologies (Lau and Botella, 2017), because it is independent to expensive instruments and friendly readout by the naked eye.Although our previous study exhibits highly specific and sensitive colorimetric LAMP to diagnosein rice, LAMP technique requires high temperature (60 ºC‒65 ºC) to amplify DNA in addition to requirement of four LAMP-primer binding sites (Buddhachat et al, 2021). TheRPA method is a cyclic DNA amplification by strand- displacement activity at low and constant temperature (30 ºC‒45 ºC) based on the enzymatic activities of proteins which are central components ofprocesses required for cellular DNA synthesis, recombination and repair (e.g., T4 gp32 as a single stranded binding protein, T4 uvsX as a recombinase, and the large fragment ofPol I as a DNA polymerase) (Piepenburg et al, 2006; Daher et al, 2016). This method requires low resource in particular energy consumption compared to LAMP. Additionally, the RPA method not only provides high sensitivity of amplified DNA detection but also demandsadditional optimization to avoid non-specific DNA amplification that oftenexhibits false-positive results in tested samples (Aman et al, 2020).
More recently, clustered regulatory interspaced palindromic repeat (CRISPR) systems have been identified in bacterial and archaeal genomes. These constitute the adaptive immune defense mechanism protecting against invading foreign nucleic acids such as plasmids and bacteriophages by cleaving and degrading their foreign DNA (Barrangou and Horvath, 2017). The CRISPRgenome-editing tool comprises twomain components as a CRISPR-RNA (crRNA) or guide RNA (gRNA) and a CRISPR-associated endonuclease (cas protein) to form a ribonucleoprotein complex (Adli, 2018). The CRISPR-cas system has been widely used as a nucleic acid detection platform to accelerate pathogen diagnosis (Chen et al, 2018; Aman et al, 2020). Nucleic acid detection based on the CRISPR- cas12a system involves two cleavage activities of the target double-stranded DNA (dsDNA)-sequence cleavage and collateral cleavage activity of non-targeted single- stranded DNA (ssDNA) (Li et al, 2018a). In the presence of the dsDNA target in the tested sample, a binary complex (cas12a and crRNA) is activated through binding the crRNA complex to a complementary dsDNA target sequence, which has a protospacer adjacent motif (PAM) (TTTV) at 5′-end and a cas12a cleave dsDNA target (Li et al, 2018b). Subsequently, the collateral cleavage activity of a ternary complex (cas12a, crRNA-targeted dsDNA-short and ssDNA) is formed, leading to cleavage of the short ssDNA probe nonspecifically (Chen et al, 2018), and then releases of the quenched fluorescent signal that indicates the presence of the target nucleic acids of interest (Creutzburg et al, 2020). The quenched fluorescent ssDNA reporter is performed as a probe to measure the formation of ternary complexes by illuminating the fluorescence signal (Li et al, 2018a). This CRISPR- cas12 technology has been successfully developed for highly specific, sensitive and fast nucleic acid detection in viral or bacterial pathogens such as African swine fever virus (Bai et al, 2019),(Xiao et al, 2020) and pathogenic bacteria in foods (Liu et al, 2021).
In this study, the RPA-assisted cas12a (RAC)system was developed as a specific, sensitive and simple method for nucleic acid detection of artificially and naturally-infected rice for on-site detection in paddy fields.
RESULTS
In vitro synthesized gRNA assessment
The gRNA-Xoo80 or gRNA-Xoo4009transcripts were separately synthesized through anreaction using DNA templates from duplex DNA ofor, respectively, driven by T7 RNA polymerase (Fig. 1-A and -B). Individual gRNA-Xoo80 and gRNA-Xoo4009transcripts were qualified by agarose gel electrophoresis and visualized by staining with novel juice. As shown in Fig. 1-C, the target bands (expected length of about 44‒60 nt) corresponding to the gRNA-Xoo80 and gRNA-Xoo4009 transcripts were highly enriched, sharp and without a smear. Moreover, the concentrations of gRNA-Xoo80 and gRNA-Xoo4009 transcripts were 400 and 190 µmol/L, respectively, as calculated by a UV spectrophotometer assay.
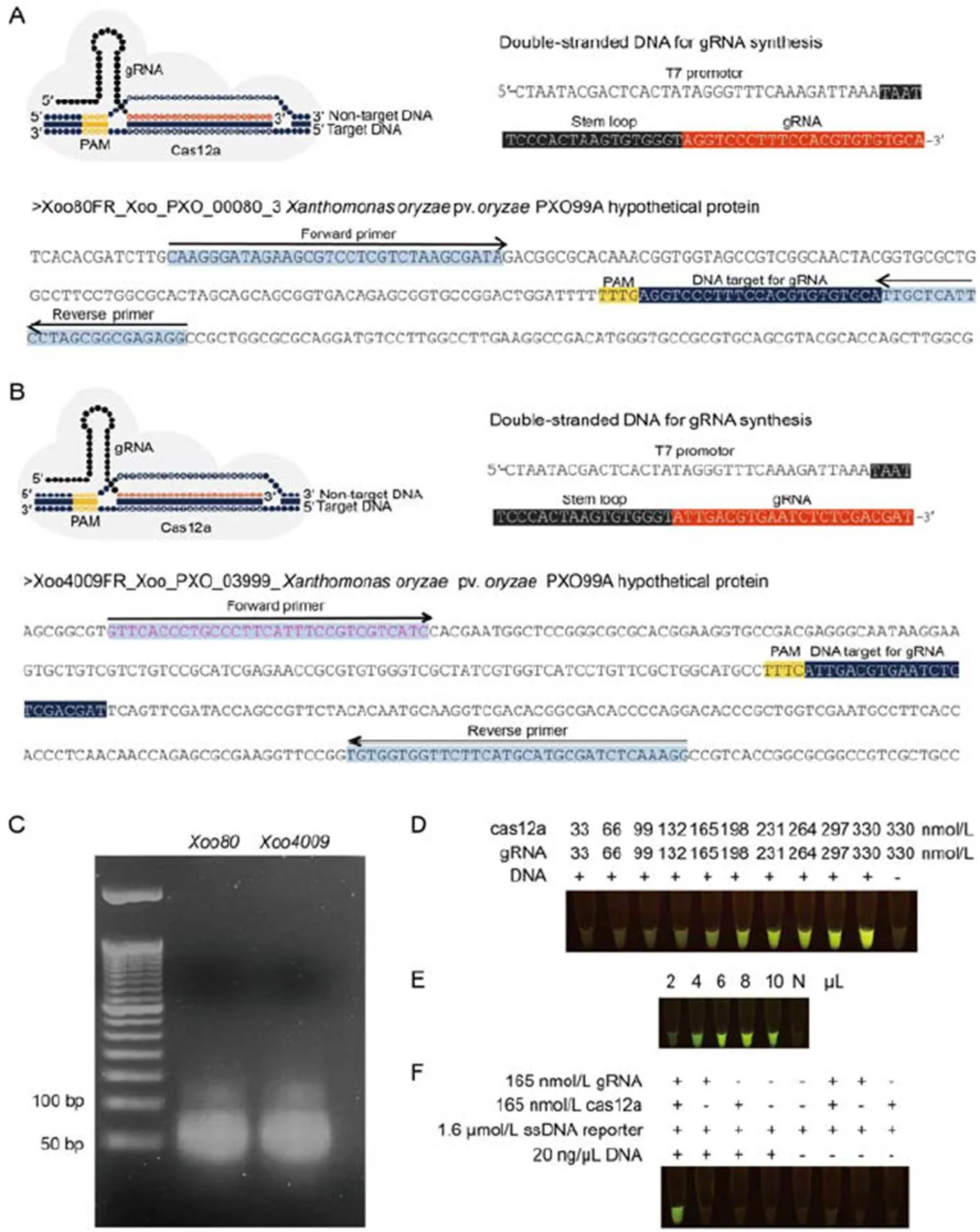
Fig. 1. Guide RNA (gRNA) synthesis andcas12a digestion.
A and B, Tertiary complexes for cas12a: gRNA:DNA targets, DNA templates for gRNA synthesis usingtranscription with T7 RNA polymerase and DNA targets containing gRNA binding sites for(A) and(B) were amplified by recombinase polymerase amplification (RPA). Protospacer adjacent motif (PAM) was used as a proto-adjacent motif.
C, Synthesized gRNA-Xoo80 and gRNA- Xoo4009 were visualized under 3% agarose gel electrophoresis.
D‒F, Experimental procedures were conducted underdigestion to assess the efficiency of concentration ratios of cas12a and gRNA varied from 33:33 to 330:330 nmol/L.
E, Sensitivity of various amounts of RPA-Xoo4009 product ranging from 2 to 10 µL. N represents negative control (without RPA product).
F, dsDNA digestibility with different chemical modifications (gRNA-Xoo4009, cas12a and RPA-Xoo4009 product).
In vitro digestion by cas12a
To optimize enzymatic cleavage activity of the RPA product, various concentrations of binary complex assembled between cpf1 (or cas12a) and gRNA- Xoo4009transcripts (as 1:1 ratio) were carried out, ranging from 33:33 to 330:330 nmol/L underdigestion reaction using RPA-Xoo4009 product and incubated at 37 ºC for 1 h. After forming the tertiary complex (binary complex and RPA product), the cas12a in this complex triggered-cleavage activities on the ssDNA reporter to emit a fluorescent signal under an LED trans- illuminator. Results showed thatthe visualized fluorescence inten- sity was dose-dependent (Fig. 1-D). The minimal concentration ratio of 165:165 nmol/L (cas12a: gRNA) was clearly observed as a strong fluorescent signal by the naked eye viadigestion (Fig. 1-D). This concen- tration of binary complex was also used to validate the sensitivity of various amounts of RPA-Xoo4009 product ranging from 2 to 10 µL underdigested product assay. Results found that the least volume of 4 µL (or 80 ng) of RPA product exhibited a detectable fluorescence signal under the LED transillumination (Fig. 1-E).
To verify the accuracy of the RAC system for-DNA detection, andigestion reaction was performed with different chemical combinations (with or without cas12a, gRNA-Xoo4009transcript, ssDNA reporter and RPA-Xoo4009 product), as shown in Fig. 1-F. Results showed that positive detection of a fluorescent signal from the digested product was only found in the treatment containing all cas12a, gRNA- Xoo4009transcript, ssDNA reporter and RPA- Xoo4009 product. However, this study did not show false positive detection from the remaining treatment combinations of the digestion reaction (Fig. 1-F).
RPA system optimization
To attain the best RPA-DNA amplification atorlocus, the RPA reaction was prepared by combining-DNA template (20 ng) with individual RPA primer pair (RPA-Xoo80 or RPA-Xoo4009) and incubating at 10 different temperatures (15 ºC‒50 ºC) for 40 min in a thermocycler. The RPA-DNA product was detected by an agarose gel electrophoresis (AGE) assay anddigestion with gRNA-cas12a binary complex activity. Results showed that the positive RPA-DNA product amplified by RPA-Xoo80 (Fig. 2-A) or RPA-Xoo4009 primer pair (Fig. 2-B) at an incubated temperature of 25 ºC‒42 ºC was clearly visualized as detectable using the AGE assay (with one target band) and fluorescent signal assay (yellowish- green color) under the LED transilluminator.
To reduce amplification time in the RPA system, seven incubation times (ranging from 0 to 60 min) were validated under the optimal condition above. Results showed that the PRA-Xoo80 primer pair (at 5 min of initial time) was quicker to amplify the target RPA-DNA product than the RPA-Xoo4009 primer pair (at 10 min). These positive products were visually detected using the AGE assay (with one target band) and fluorescent signal assay (yellowish-green color) (Fig. 2-C and -D). The optimal condition of RPA system was performed at 39 ºC for 30 min with bothandloci.
Specificity and sensitivity determinations of RAC system
The appropriate condition was used to evaluate the specificity and sensitivity of both the RPA (at 39 ºC for 40 min) and RAC systems (at 37 ºC for more than 60 min), compared to the standard PCR system. To determine the specificity of the RPA and RAC systems fordetection, the RPA primers and gRNA, designed from the internal regions of the two specificloci (and), were validated fordetection using the PCR, RPA and RAC systems. Ten bacterial species comprised of the standard-SK2-3, threespecies (,and) andsix other bacterial genera (,,,,and) (Table S1), were cultured in nutrient broth (NB) medium. Their DNAs were separately extracted to use as DNA templates (20 ng) for all the tested assays. The RPA-Xoo80 primer pair in the PCR assay generated a positive PCR product (~200 bp) in both bacterialandspecies (Fig. 3-A). Meanwhile, the RPA primer and gRNA sequence of thelocus were precisely specific for bacterialspecies, as shown by positive RPA amplification (~302 bp) using the AGE technique and digested-RPA product with strong fluorescent signal under the LED transilluminator (Fig. 3-B). This suggested that the RPA and RAC systems of thelocus were highly specific fordetection compared to the PCR system. The RPA primer and gRNA of thelocus were less specific in all the tested bacteria than thelocus because they amplified similar profiles of positive RPA products (~200 bp) and digested-RAC products inandspecies through visual detection by the AGE assay and fluorescent signal assay, respectively (Fig. 3-A). Moreover, the remaining bacterial species and genera were absent for PCR, RPA and digested-RAC product detection for all assays (Fig. 3-A).
To determine the sensitivity of the RAC system fordetection atandloci, the experiments were performed under two conditions of (i) various numbers of target-DNA templates (0‒1 × 108copies/µL), and (ii) various numbers of bacterial cells (0‒2.5 × 108cfu/mL). Results showed that the RPA primer and gRNA sequence of thelocus generated positive RPA products and cleavage products, requiring reduced limit of detection (LOD) numbers of DNA templates (1 × 102copies/µL) (Fig. 3-C) and bacterial cells (2.5 × 103cfu/mL) (Fig. 3-E). These consistently resulted in product detection by the AGE and fluorescent signal techniques.

Fig. 2. Optimization of recombinasepolymerase amplification (RPA) reaction for RPA-DNA amplification based on Xoo80 or Xoo4009 primer pairs.
A and B, RPA was performed under various temperatures ranging from 15 ºC to 50 ºC for RPA-Xoo80 (A) and RPA-Xoo4009 (B) primer pairs.
C and D, Time duration for DNA amplification was conducted at different periods of 0‒60 min for RPA-Xoo80 (C) and RPA-Xoo4009 (D) primer pairs after obtaining the optimal temperature of 39 ºC. N represents negative control which was the reaction without DNA templates.
Fig. 3. Specific and sensitive assessments of RAC system fordetection compared to RPA and PCR systems.
A and B, Specificity test of RAC system using(A) andloci (B) was performed with differentspecies and other bacterial species.
C‒F, Sensitivity test of RAC system using(C and E) andloci (D and F) was determined with DNA templates from plasmid DNA (pUC57-Xoo80 or pUC57-Xoo4009) ranging from 0 to 1 × 108copies/µL (C and D), and bacterial number ranged in 0‒2.5 × 108cfu/mL (E and F).
RPA, Recombinase polymerase amplification; RAC, RPA-assisted cas12a. N refers to an RPA reaction without DNA templates as a negative control.
By contrast, the RAC system using the RPA primer and gRNA sequence of thelocus was less sensitive than thelocus because LOD required the minimal number of DNA templates at 1 × 103copies/µL (Fig. 3-D) and bacterial cells at 2.5 × 103cfu/mL (Fig. 3-F) for thelocus. However, theleast numbers of DNA templates (1 × 105copies/µL) and bacterial cells (2.5 ×105cfu/mL) were visualized bydetection using thelocus for RPA by AGE. For all the assessmentsof specificity and sensitivity of the RAC system using the both loci, thelocus gave superior specificityperformance over thelocus. Thus, thelocus was chosen fordetection under the RAC system.
Compatibility determination of optimal RAC system
The optimal RAC system for both specificity and sensitivity fordetection was carried out undercondition of DNA templates (1 × 105copies/µL) with the RPA-Xoo4009 primer for RPA amplification system (at 39 ºC for 40 min) and binary complex formation of cas12a and gRNA-Xoo4009 sequence for the RAC digestion system (at 37 ºC for 60 min). To examine the compatibility of the RAC system fordetection, 11 different bacterialisolates (K1E, K2, M6, K4G, E, K4C, M2, F, D, K1G and SK2-3) listed in Table S1 were cultured in NB medium, with DNA extracted to use as templates in the tested system. Results displayed that all the testedisolates successfully amplified the positive RPA-DNA product and were able to detect target band (~302 bp) by the AGE assay (Fig. 4-A). Similarly, they also generated positive cas12-digested product, which were able to be visualized as fluorescence signal (yellowish-green color) by the RAC assay (Fig. 4-B) and quantified fluorescent intensity (8 000‒15 000 AU) by a real-time PCR machine (Fig. 4-C). This strongly confirmed that the RAC system using thelocus was feasible to detectin endemic areas.
RAC system for Xoo detection in artificially infected rice
To further evaluate the sensitivity of the optimal RAC system under different bacterial densities, leaf samples of rice variety RD47 were artificially infected withcell density ranging from 25 to 2.5 × 108cfu/mL, and collected at 7 d post-inoculation (dpi) for plant crude extraction. After 7 dpi, theartificially infected leaf samples with bacterial cell density of 2.5 × 108cfu/mL appeared obviously typical BLBdisease symptoms such as wilted and rolling leaves in infected regions, whereas the other infected bacterial densities displayed no BLBdisease symptoms (Fig. 5-A). Results of the RAC assay showed increased sensitivity to detectthan the disease symptom assay because the positive fluorescent signals enabled detection in samples infected with low bacterial cell density of 2.5 × 103cfu/mL (Fig. 5-B and -C).
To further evaluate the sensitivity of the optimal RAC system at different times, leaf samples of rice variety RD47 were artificially infected withcell density of 2.5 × 108cfu/mL and collected at 0‒28 dpi for plant extraction. The cleaved product was visually detected under a LED transilluminator and quantified for fluorescent signals by a real-time PCR assay. Results showed that BLB lesions were clearly diagnosed at 7 dpi (Fig. 6-A), whereas the RAC system gave positive fluorescence signals from 3 dpi (Fig. 6-B and -C). This indicated that the RAC assay was more sensitive at early bacterial infection than phenotypic BLB observation.
Fig. 4. Compatibility test of optimal RAC system usinglocus.
Detection feasibility of RAC used 11isolates (K1E, K2, M6, K4G, E, K4C, M2, F, D, K1G and SK2-3) distributed in the endemic area. The resulting products were visualized by agarose gel electrophoresis (A) and an LED transilluminator (B), and the fluorescence measurement was quantified using a real-time machine (C). N refers to an RPA reaction without DNA templates as a negative control. RPA, Recombinase polymerase amplification; RAC, RPA-assisted cas12a.
Application of RAC assay for Xoo detection in natural rice samples
In this study, the RAC assay was employed to detectin natural rice samples collected from different fields in the northern of Thailand (Table S2). Twenty- three rice leaf samples with visible BLB symptoms were used for plant extraction under pre-heating and adding EDTA. Their extracted samples were used for RPA-DNA amplification by the optimal RPA assay and then subjected todigestion by the optimal RAC system. Of these, 17 samples showed positive results fordetection by exhibiting fluorescent signals (Fig. 7-A and -C), while only 10 of the 17 samples showed positive RAC results of amplified DNA product using the traditional PCR assay (Fig. 7-B). This finding indicated that the developed RAC assay provided higher sensitivity fordetection in naturally infected rice samples than the conventional PCR assay.
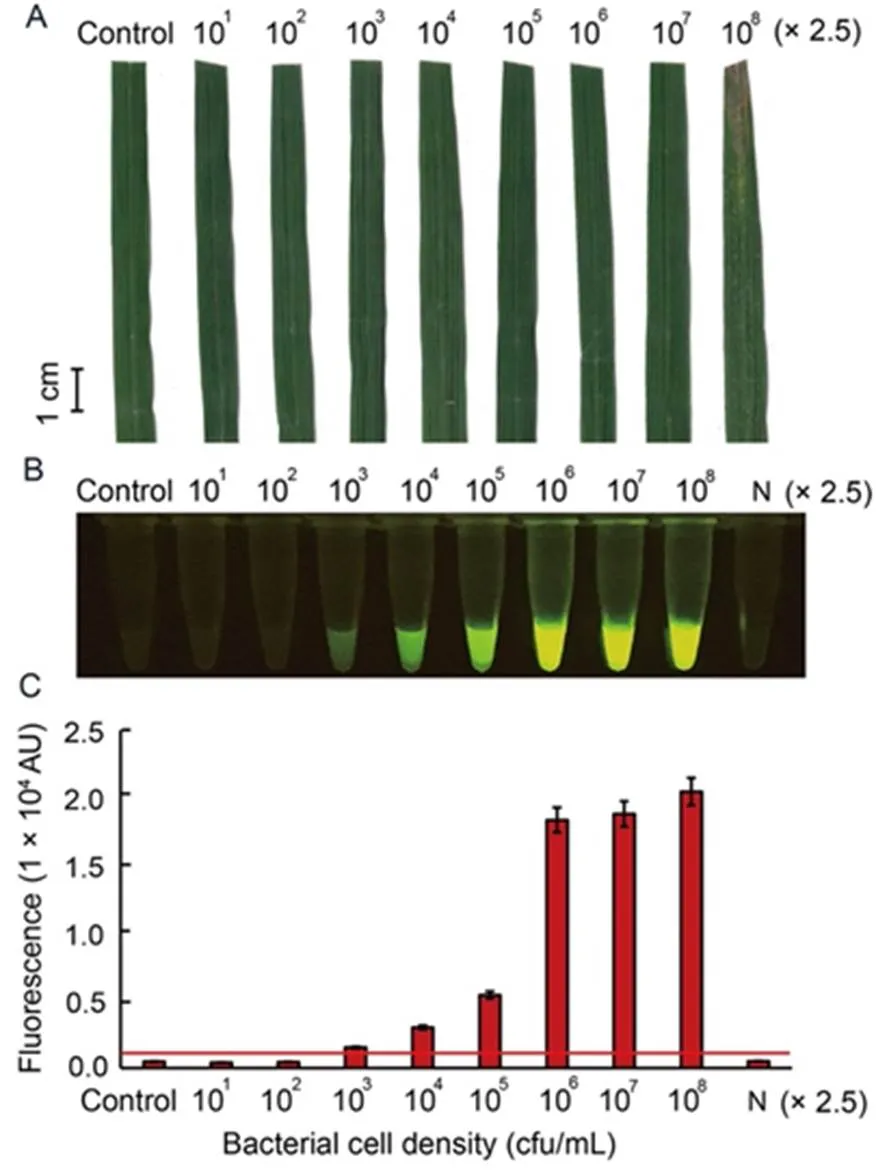
Fig. 5. Sensitivity of RAC system fordetection in artificially infected rice leaf, RD47 variety, with different bacterial cell intensities.
Sixty-day-old rice leaves were inoculated withat different concentrations ranging from 25 to 2.5 × 108cfu/mL for7 d post- inoculation. External bacterial leaf blight symptoms were photographed (A). Leaf extracts were used as DNA templates in anRAC system and fluorescent signals from the RAC products were visualized by an LED transilluminator (B), and quantified by a real-time PCR machine (C). N refers to an RPA reaction without DNA templates as a negative control. RPA, Recombinase polymerase amplification; RAC, RPA-assisted cas12a.
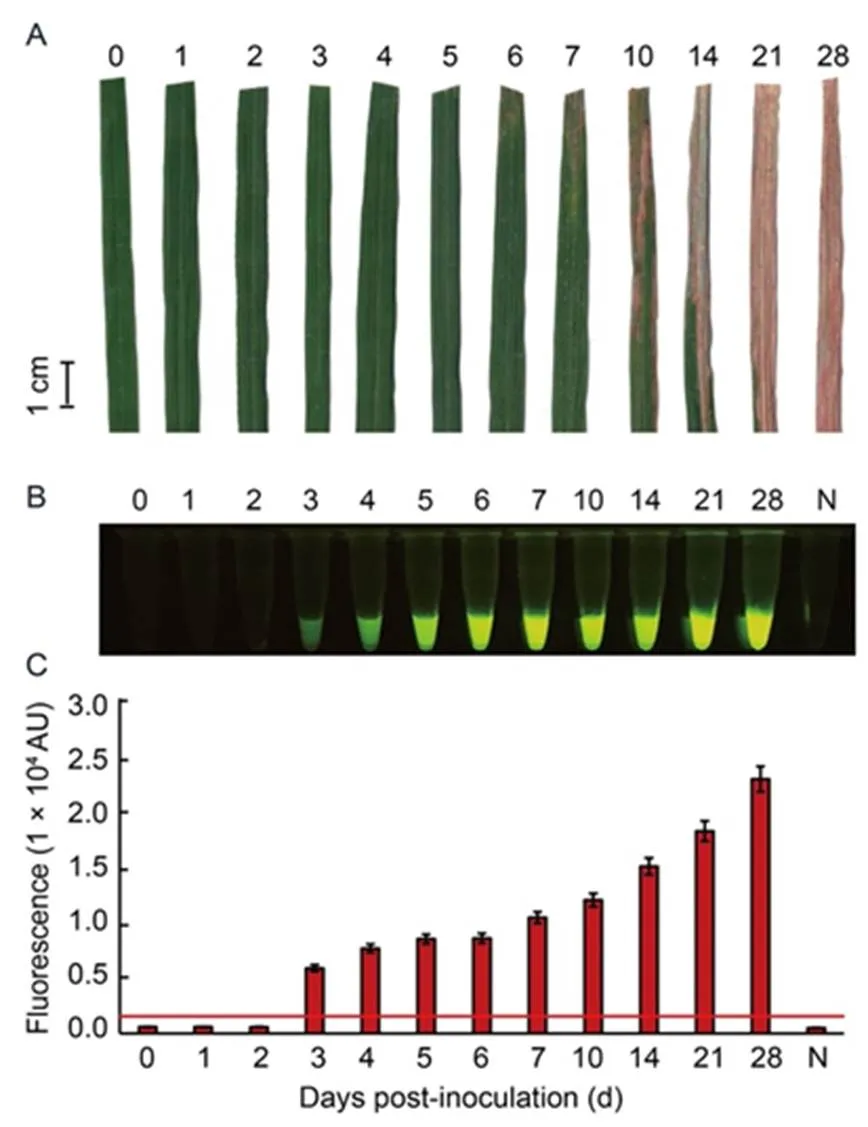
Fig. 6. Sensitivity of RAC system fordetection at different days post-inoculation (dpi) of rice leaves.
Leaf samples of rice variety RD47 were artificially inoculated withat 2.5 × 108cfu/mL by the clipping method. Leaves were collected at different time courses of 1–28 dpi fordetection. External bacterial leaf blight symptoms were photographed (A). Leaf extracts were used as DNA templates in anRAC system and fluorescent signals from the RAC products were visualized by an LED transilluminator (B), and quantified by a real-time PCR machine (C). N refers to an RPA reaction without DNA templates as a negative control. RPA, Recombinase polymerase amplification; RAC, RPA-assisted cas12a.
DISCUSSION
Despite comprehensive control strategies to prevent the spread of disease in rice fields, the bacterialpathogen has continued to decimate rice productivity in Thailand. Bacterial-specific diagnosis of infected plants is crucial for early detection and containment. Recently, integration of RPA and CRISPR-cas12a techniques has successfully increased sensitive, specific and portable detection of nucleic acids in various pathogens including viruses in shrimp (Chaijarasphong et al, 2019), Methicillin-resistant(Xu L Q et al, 2020),(Ai et al, 2019),(Kanitchinda et al, 2020), SARs-CoV-2 (Ali et al, 2020), viruses in plants (Aman et al, 2020) and food (Liu et al, 2021). This new integrated system can easily harness nucleic acid detection at a single temperature, thus supporting practical use in the field for on-site diagnosis without the need of well-equipped laboratories (Liu et al, 2021).
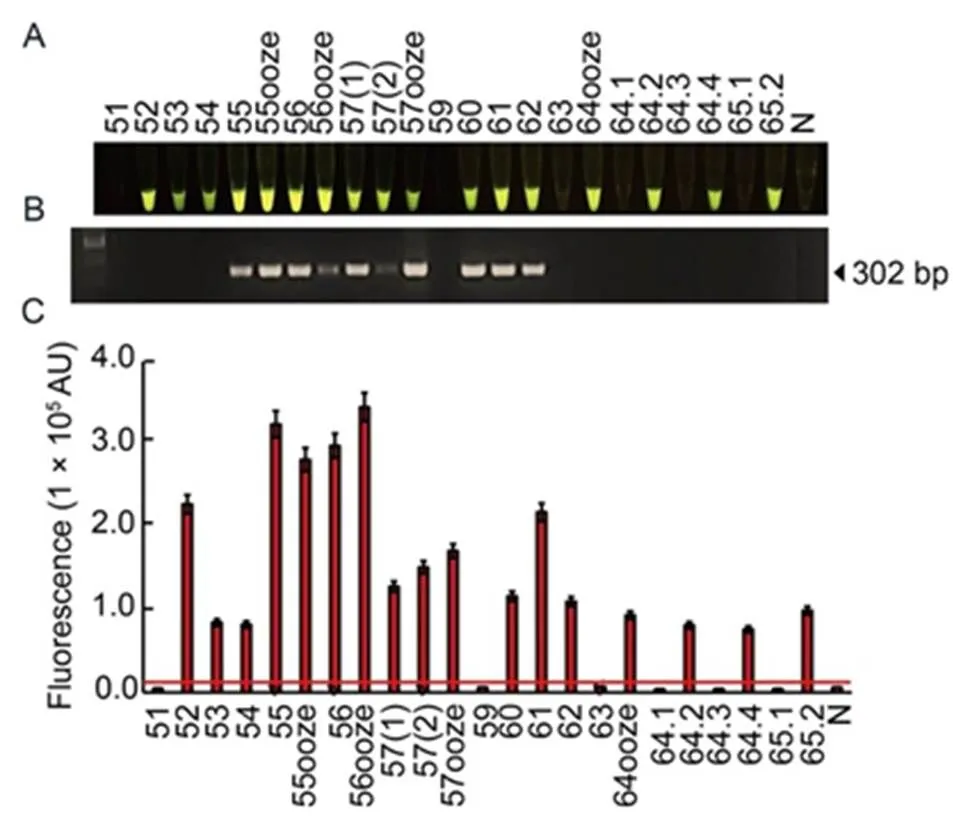
Fig. 7. Diagnosis ofpathogen using RAC system in naturally infected rice samples.
A total of 23 leaf samples were collected from rice fields in lower northern of Thailand. Plant extracts were used for DNA templates in the RAC and PCR reactions. The cas12a-mediatedcleavage fluorescence signals from the RAC assay were detected under an LED transilluminator (A), PCR products were visualized by agarose gel electrophoresis (B), and their intensities were quantified by a real-time PCR machine (C). N refers to an RPA reaction without DNA templates as a negative control. RPA, Recombinase polymerase amplification; RAC, RPA-assisted cas12a.
Here, the RAC system was developed fordetection in infected rice leaves. In the RAC system, the target RPA-DNA amplification step of bacterialserves as a substrate for cas12aactivity of the tertiary complex (cas12a-gRNA-dsDNA target) (Li et al, 2018b). The cas12a targeting of the dsDNA triggers its collateralactivity that cleaves the ssDNA reporter molecules and releases a fluorescence signal that can be visualized by an LED transilluminator (Ding et al, 2020). Our findings exhibited that the RAC system using thelocus had high efficiency to specifically detectwithout cross- reaction of other relating species or genera, and high sensitivity with less LOD of DNA concentration (1 × 103copies/µL) and bacterial cell density (2.5 × 103cfu/mL). This RAC system also specifically diagnosed 11species isolated from epidemic areas in lower northern Thailand. The RAC system was appropriate for implementation indiagnosis of artificially and naturally infected rice using direct plant extracts (without DNA isolation).
In vitro digestion of cas12a system
In this study, abundant gRNA-Xoo80 and gRNA- Xoo4009 RNA molecules were generated viatranscription using synthetic dsDNA as a template.These results agreed with previous findings that an-transcribed system is beneficial to produce ssRNA transcripts, triggered by the T7 promoter sequence annealed to a forward-directed T7 promoter primer that activates T7 RNA polymerase activity (Liu et al, 2019; Aman et al, 2020). Optimal enzymatic activity of the binary complex was determined byformation between gRNA transcript and cas12a ribonuclease in the optimal ratio of 165:165 nmol/L. Results concurred with Zetsche et al (2015) who suggested the optimal ratio of 1:1 (cas12a:gRNA) as suitable for binary complex formation. Similarly, Mahas et al (2021) reported that a concentration ratio of cas12a:gRNA (at 250:250 nmol/L) is required for the cas12-based detection system. Results of thedigested RAC system implied that the tertiary complex (cas12a-gRNA-dsDNA target) had two enzymatic activities. The cas12a-basedactivity firstly cleaved the dsDNA target of the RPA product, followed by activating the cas12a-basedactivity to digest the ssDNA fluorescent quencher reporter, causing the exposure of fluorescence signals as positive results, as supported by Li et al (2018b) and Ding et al (2020).
RAC performance for Xoo detection
To optimize the RAC detection system, eithergRNA- Xoo80 or gRNA-Xoo4009 transcript was examined for sensitivity and specificity ofdetection, compared to the traditional PCR assay. Results showed that the RAC system using gRNA-Xoo80 transcriptposed to be more sensitive than gRNA- Xoo4009, corresponded to 10 and 100 times when using DNA templates from different amounts of DNA copies and bacterial cells, respectively. This might be that the RPA-Xoo80 amplicon (~200 bp) was smaller than the RPA-Xoo4009 amplicon (~300 bp), which was performed with shortened incubation times during cyclic DNA amplification to generate massive amount of RPA products, used as substrates for triggeringcas12a digestion. By contrast, the RAC system using thelocus was highly specific for onlydetection, while the RAC system using thelocus generated cross-reaction of RPA amplification and RAC-digested products in bothandspecies. Moreover, compatibility of the RAC system with thelocus consistently amplified the positive target products in 11 testedisolates identified from some lower northern areas of Thailand. These results also agreed with previous findings that colorimetric LAMP (cLAMP) system based onlocus gives more sensitivity but less specificity thanlocus for detectingin rice leaves (Buddhachat et al, 2021). One possible explanation of these results was that thelocus contains unique nucleotide sequences found only in thestrain, whereas thelocus shares nucleotide sequence similarity with extrachromosomal DNA of two otherspecies includingpv.CFBP 5530 () plasmid pXap41 (GenBankaccession number FR875157) andpv.306 () plasmid pXAC33 (GenBank accession number AE008924) (Lang et al, 2014). However, these two strains have not been reported as infecting rice. Therefore, to prevent the risk of false positives from other pathogens possibly contaminated from the environment, thelocus was consistently suitable for-specific detection by the RAC system. In comparative sensitivity oflocus fordetection, this developed RAC assay posed to be more sensitive (required as little as 1 × 103cfu/mL) than cLAMP usinglocus (required as little as 1 × 105cfu/mL) (Buddhachat et al, 2021). Here, the RAC assay considerably trended to be more sensitive than the LAMP assay. Moreover, the RPA method uses less energy to perform DNA amplification than the LAMP assay, suggesting it can be a promising tool for field-deployable detection.
Xoo detection by RAC without DNA extraction
The capability of RAC system was developed to simplifydiagnosis from the direct-rice-leaf extraction without-DNA isolation. Here, we validated the utility of RAC system using thelocus to determine the presence ofusing rice leaf tissue extracted by adding EDTA (0.8 mmol/L) and pre-heating with high temperature (98 ºC for 10 min). This treatment combination enables to eliminate inherent DNase activity from rice leaf extracts, because DNase contamination has activity to digest ssDNA reporters to produce the fluorescence signal, causing a false positive determination. These findings are supported by observations that the EDTA chelating agent and high temperature play an important role to completely inhibit DNase activity contaminated in the plant extracts (Barra et al, 2015), and reduce false positive detection from digested- ssDNA reporter by DNase (Rudresha et al, 2019). Thereby, additional EDTA and pre-heat are highly necessitated to assaydetection from rice leaf extracted by the RAC system.
Field-deployable Xoo detection by RAC
In the field test, the RAC assay was validated to detectin the infected rice samples. Rice variety RD47 was artificially infected by the clipping method with(2.5 × 108cfu/mL). The RAC assay based on thelocus had clearly facilitateddiagnosis at early infection (3 dpi) before BLB symptomatic appearance on their leaves. Furthermore, this developed RAC system was applied to diagnosein naturally infected rice leaf samples collected from rice fields in lower northern Thailand. The RAC assay provided better performance ofdetection as higher numbers of positive samples, compared to the PCR assay, indicating the higher sensitivity of the RAC method. In agreement with previous finding, the RPA-cas12a- based system is highly valuable and sensitive for on-site detection of foodborne pathogenic bacteria (Liu et al, 2021).
Therefore, this RAC assay provided feasible point- of-care (POC) testing forsurveillance in fields and long-term BLB disease management to reduce rice-yield product. Afterdetectionof infected rice leaves in fields at the early stage, we suggested to spray muriate of potash (MOP) (6 g/L), Zn fertilizer (4 g/L) and elemental sulphur (6 g/L) or apply additional MOP (37.5 kg/hm2) to protect rice fromattack, because potassium in MOP significantly promotes the thickness of outer cell wall, thereby increasing the resistance ofattack and spread (Ansari et al, 2020). Moreover, to prevent thetransmission from BLB-infected rice fields to the vicinity of BLB-noninfected rice fields, we suggested that rice cultivation infected with BLB must be diminished, then drained out water, and dried for 7‒10 d to eliminate thefrom the field (Ansari et al, 2020).
Limitation of Xoo detection by RAC
A limitation of RAC assay to diagnosewas the complexity in its process, which was consisted of two separating steps: (i) DNA amplification by RPA procedure, and (ii)cleavage of DNA target by cas12a, leading to possible contamination in digestion of DNA target, resulting in the false positive. Thus, one-tube assay should be developed to integrate the amplification and detection steps into a single reaction for simplicity and reduction of contamination risk (Aman et al, 2020; Wang et al, 2020). Although the current cost of RAC system is more expensive, approximately 1 $ per reaction, than the conventional PCR, these were affordable and reasonable to gain more straightforward and robustness method for POC testing (Gootenberg et al, 2017; Zhang et al, 2020).
The RAC system is highly sensitive and specific with rapid detection of bacterial, with or without the DNA extraction step, and can be implemented at POC diagnosis in rice fields. The RAC assay is simple and only requires a heat box and an LED transilluminator, which are cost-effective and portable (Liu et al, 2021). In agreement with recent findings, the cas12a-based system has been developed for pathogenic diagnoses at POC applications worldwide, such as papilloma virus-16 and 18 (HPV16 and HPV18) in human (Li et al, 2018a), white spot syndrome virus in shrimp (Sullivan et al, 2019), tuberculosis bacteria in human (Xu H P et al, 2020) and pathogenic bacteria in food (Liu et al, 2021). The read-out fluorescent signal is dependent on the digested ssDNA reporter by cas12a activity. This is beneficial to modify further as the lateral flow dipstick (LFD) method, which would be the best choice for implementing POC diagnosis in rice fields because the positive products can be easily read by the naked eye from the band on the strip (Wang et al, 2020).
The RAC system is established for improving performance to acceleratedetection in the field with high sensitivity, specificity, robustness and simplicity. This system enables diagnosis of bacterialfrom infected rice leaves with no requirement for DNA isolation and sophisticated instruments. This optimal RAC system offers a promising powerful tool fordetection at the POC application. In addition, we believe that this method would be useful as a quarantine tool for epidemiological studies ofpresence in fields in both rice and its environmental samples (i.e., soil and water) for rapid and precise surveillance and management ofoutbreaks in rice fields to reduce yield loss.
METHODS
Bacterial species
Twenty-one bacteria used in this study (Table S1) consisted of 12isolates (K1E, K2, M6, K4G, E, K4C, M2, F, D, K1G, PPRDO and SK2-3), 3genera includingpv.(),pv.(),pv.(), and 6 bacterialspecies(),(),(),(),() and(). From the 12isolates, 10 differentisolates identified by-specific primers (Lang et al, 2010) were collected from cultivated rice areas in the lower northern region of Thailand. There were five standardspecies characterizedby Carpenter et al (2018), the Plant Protection Research and Development Office (PPRDO), or the Thailand Institute of Scientific and Technological Research of Thailand (TISTR) (Table S1).
DNA extraction
For bacterial DNA extraction, individual bacterialspecies were cultured on NB medium (peptone 10 g/L, beef extract 3 g/L and NaCl 5 g/L, pH 7.0). The bacteria were incubated at either 28 ºC for 72 h (forspecies) or 37 ºC for 24 h (for other bacterial species), with shaking at 180 r/min until OD600reached approximately 0.2 (cell density of 2.5 × 108cfu/mL). Bacterial cell pellets were collected by centrifugation at 5 000 r/min for 10 min, and DNA was extracted using a Genomic DNA Isolation Kit (Blood/Cultured cell/Fungus) (GeneDireX, Taiwan, China), according to the manufacturer’s instructions. The extracted DNA sample was quantified and qualified at OD260and OD280using a spectrophotometer (Nabi-UV/Vis Nano Spectrophotometer, MicroDigital, South Korea)and 1.0% agarose electrophoresis assay to examine DNA integrity. The DNA was stored at -20 ºC until further use.
RPA procedure
Lang et al (2010) reported that two specific primer pairs (Xoo80-F/R and Xoo4009-F/R) designed from PXO_00080 and PXO_03999 loci, respectively, can successfully detectedDNAstrain PXO99A (CP000967.2). In this study, the sequences of these loci obtained from the NCBI database were used to manually design the forward and reverse RPA primer pairs. An individual RPA primer, corresponding to 30‒33 bp length, was generated by adding more complementary nucleotides to its DNA template at the 3′-end of the RPA- Xoo80-F/R or RPA-Xoo4009-F/R primers (Fig. 1-A and -B). The primer sequences are shown in Table S3. Each RPA-F/R primer pair was determined for specificity toDNA using the RPA system with TwistAmp Basic Kit (TwistDx, UK). According to the instruction manual, reagents for the RPA reaction (25 µL) in the PCR tube consisted of 12.5 µL of 2× reaction buffer, 1.8 mmol/L of dNTPs, 5 µL of 10× basic E-mix, 0.48 µmol/L of RPA primer, 1.25 µL of 20× core reaction mix, 14 mmol/L of magnesium acetate, 20 ng of-DNA template, and final volume adjusted with nuclease- free water. The mixture was incubated at 39 ºC for 40 min. Positive RPA-DNA amplifications were examined by 3.0% agarose gel electrophoresis assay.
Design of dsDNA for gRNA synthesis
The specific gRNA oligonucleotides (23‒25 nt) were designed within nucleotide sequences of the DNA region amplified by RPA using RPA-Xoo80-F/R or RPA-Xoo4009-F/R primers under the requirement of PAM sequence (5′-TTTN-3′) at the 5′-end of gRNA (Chen et al, 2020). The dsDNA fragment was constructed by the Integrated DNA Technologies (Coralville, USA). The map position of oligonucleotide fororcontained three main components of T7 promoter region for driving transcription, crDNA sequence (21 nt) and gRNA sequence (23 nt) (Fig. 1-A and -B). The crRNA sequence contained two compartments including stem loom at 5′-end to form a binary complex with cas12a endonuclease and the gRNA sequence which is complementary base to bind to the target-DNA template.
In vitro gRNA transcript
Synthetic dsDNA templates fororwere separately used to transcribe the gRNA molecules undertranscription, driven by the T7 promoter of HiScribe™ T7 Quick Kit (E2050S, New England Biolabs Inc, USA). The reaction system (30 µL) contained dsDNA foror(2 µg) used as dsDNA templates and 2 µL of T7-RNA polymerase, and final volume was adjusted with nuclease-free water. The mixture was incubated at 37 ºC for 16 h (overnight) under a thermocycler (Thermo Fisher Scientific, USA). The gRNA transcript was synthesized according to the protocol with slightly modified from Liu et al (2019).
The gRNA transcripts were purified and cleaned to removethe incorporated NTPs using the Monarch RNA Cleanup Kit (New England Biolabs Inc, USA). The purified product was qualified and quantified at OD260and OD280using a UV spectrophotometer (Nabi-UV/Vis Nano Spectrophotometer, MicroDigital, South Korea), while integrity was examined by the 3.0% agarose gel electrophoresis assay.
RAC system
RAC system fordetection was established by following four steps: (i) The positive RPA-DNA product, amplified by either RPA-Xoo80 or RPA-Xoo4009 primer pair, served as a substrate for the cas12aactivity of the tertiary complex (cas12a-gRNA-target dsDNA) underdigestion. Cas12a or cpf1 (EnGen®Lba Cas12a, M0653T) was purchased from New England BioLabs Inc, USA. This determined the performance of the RAC system underdigestion of cas12a assembled with specific gRNA for DNA target of eitherorlocus. (ii) The binary complex of gRNA transcript and cas12a ribonuclease (cpf1) was formed in the RAC reaction (24 µL) containing 3 µL of 10× NEB buffer2.1, 165 nmol/L of cpf1 or cas12a ribonuclease and 165 nmol/L of gRNA transcript of eitheror. The mixture was incubated at 37 ºC for 10 min. (iii) The ssDNA reporter labeled with 5′FAM-3′IABkFQ (1 µL of 50 µmol/L) and RPA-DNA product (5 µL) used as dsDNA substrate for cas12a were added into the mixture and inverted 10 times. Nuclease-free water was used to adjust final volume and the mix was incubated at 37 ºC for 60 min. (iv) The cas12a-mediated cleavage of ssDNA reporter (collateralactivity) was detected by visualizing the florescence signal emission with the naked eye under an LED transilluminator (GeneDireX, Taiwan, China). The negative amplification product withoutDNA template from the RPA-DNA reaction was used as a blank control.
For RPA amplification using the DNA template from crude plant extracts, the RPA reaction must be carried out by heating (at 98 ºC for 10 min) and adding EDTA (0.8 mmol/L) to inhibit contaminated DNase activity (File S1). DNase contamination affects the cleavage activity of the ssDNA reporter, leading to fluorescence signal emission and causing false positive detection.
Performance of RAC system
Performance of the RAC system with gRNA of eitherorlocus was examined for specificity and sensitivity ofdetection. For specificity detection, a standardSK2-3strain (Carpenter et al, 2018) and individually extracted DNA from three bacterial species and five bacterial genera (Table S1) was used as templates. The digested product was visualized for fluorescence signal emission under an LED transilluminator (GeneDireX, Taiwan, China) by the naked eye and quantified for fluorescence signal by a real-time PCR machine (Bioer, Hangzhou, China).
To assess the sensitivity of the RAC system, the plasmid concentration of pUC57-Xoo80 or pUC57-Xoo4009 construct was determined using a UV spectrophotometer and calculated for the number of plasmid copies. The plasmid solution was diluted in TE buffer to 10-fold serial concentration ranging from 1 to 1 × 108copies/µL, and this served as the detection sensitivity of the RAC system with specific gRNA of eitherorlocus. The digested product was inspected by the naked eye under an LED transilluminator (GeneDireX, Taiwan, China), and the fluorescence signal was recorded by a real-time PCR machine (Bioer, Hangzhou, China).
Consistent performance of the optimal RAC system (for both specificity and sensitivity) was further examined for compatibility by detection of a standardstrain SK2-3 and 10isolatescollected from the endemic region in Phitsanulok and Phichit provinces, Thailand (Table S1) using the same above- mentioned method.
RAC-system detection of Xoo artificially infected rice
The RAC system using gRNA-Xoo4009 gave the best performance for both specificity and sensitivity fordetection using the extracted DNA templates. This optimal RAC system was further applied fordiagnosis in artificially infected rice leaf samples with and without the DNA extraction process. The susceptible rice variety RD47 was naturally grown under greenhouse condition for 60 d at the Department of Biology, Faculty of Science, Naresuan University, Thailand, while bacterialisolate, causing severe BLB symptom, was cultured on NB medium. Sixty-day-old rice leaves were experimentally challenged with 10-fold serial dilution ofcell suspension ranged from 25 to 2.5 × 108cfu/mL, following the leaf clipping method (Yuan et al, 2016). The negative rice leaf control was clipped with the NB medium without bacterialsuspension. At the end of different time courses (0‒7 dpi), a leaf region below the clipping lesion (approximately 5 cm) was harvested, chopped into small pieces, placed into sterilized distilled water (100 µL) and incubated at 95 ºC for 10 min. The supernatant (2 μL) of crude plant extracts was used fordetection by the optimal RAC system using gRNA-Xoo4009 with adding EDTA (0.8 mmol/L) to prevent the digested ssDNA reporter with contaminated DNase reducing the false positive (File S1). The performance efficiency of the RAC system was compared to the traditional PCR assay using the Xoo4009-F/R primer pair.
RAC system application for Xoo detection in natural rice sample
After assessment of the RAC system, its performance was also applied to diagnosenaturally infected rice leaves that exhibited BLB symptoms. Samples were collected from rice fields in the lower northern of Thailand including Kamphaeng Phet, Phitsanulok, Sukhothai and Phichit provinces (Table S2). Rice leaf surfaces were cleaned with detergent and washed in tap water to eliminate bacterial contamination. The DNA template of this plant extract was used fordetection using the RAC and PCR assays, according to the method described above.
ACKNOWLEDGEMENTS
This study was financially supported by the Agricultural Research Development Agency (Public Organization), Thailand (Project No. PRP6205031190). We also thank Phitsanulok Rice Research Center, Wangtong, Phitsanulok, Thailand for providing the rice materials and Dr. Suradet Palawisut for valuable suggestion.
SUPPLEMENTAL DATA
The following materials are available in the online version of this article at http://www.sciencedirect.com/journal/rice-science; http://www.ricescience.org.
File S1. Materials, methods and results of heat and EDTA on inhibition of inherent DNase from rice leaf extracts.
Table S1. Bacterial species used for establishing RPA-assisted cas12a assay.
Table S2. Locality, stage of rice that leaf tissues were collected from agricultural area of lower northern of Thailand fordetection by RPA-assisted cas12a assay (RAC) and PCR assays.
Table S3. Oligonucleotide sequences used to amplifyby RPA method.
Adli M. 2018. The CRISPR tool kit for genome editing and beyond., 9(1): 1911.
Ai J W, Zhou X, Xu T, Yang M L, Chen Y Y, He G Q, Pan N, Cai Y W, Li Y J, Wang X R, Su H, Wang T, Zeng W Q, Zhang W H. 2019. CRISPR-based rapid and ultra-sensitive diagnostic test for., 8(1): 1361‒1369.
Ali Z, Aman R, Mahas A, Rao G S, Tehseen M, Marsic T, Salunke R, Subudhi A K, Hala S M, Hamdan S M, Pain A, Alofi F S, Alsomali A, Hashem A M, Khogeer A, Almontashiri N A M, Abedalthagafi M, Hassan N, Mahfouz M M. 2020. iSCAN: An RT-LAMP-coupled CRISPR-Cas12 module for rapid, sensitive detection of SARS-CoV-2., 288: 198129.
Aman R, Mahas A, Marsic T, Hassan N, Mahfouz M M. 2020. Efficient, rapid, and sensitive detection of plant RNA viruses with one-pot RT-RPA-CRISPR/Cas12a assay., 11: 610872.
Ansari T H, Haque S S, Uddin A. 2020. Pest management decision guide: Green and yellow list: Management of bacterial leaf blight (BLB) of rice. Plantwise knowledge Bank, CAB International. [2 October, 2021]. https://www.plantwise.org/KnowledgeBank/ pmdg/20167800391.
Bai J, Lin H S, Li H J, Zhou Y, Liu J S, Zhong G R, Wu L T, Jiang W F, Du H L, Yang J Y, Xie Q M, Huang L Z. 2019. Cas12a-based on-site and rapid nucleic acid detection of African swine fever., 10: 2830.
Barra G B, Santa Rita T H, de Almeida Vasques J, Chianca C F, Nery L F A, Santana Soares Costa S. 2015. EDTA-mediated inhibition of DNases protects circulating cell-free DNA fromdegradation in blood samples., 48(15): 976‒981.
Barrangou R, Horvath P. 2017. A decade of discovery: CRISPR functions and applications., 2: 17092.
Buddhachat K, Ritbamrung O, Sripairoj N, Inthima P, Ratanasut K, Boonsrangsom T, Sujipuli K. 2021. One-step colorimetric LAMP (cLAMP) assay for visual detection ofpv.in rice., 150: 105809.
Carpenter S C D, Mishra P, Ghoshal C, Dash P K, Wang L, Midha S, Laha G S, Lore J S, Kositratana W, Singh N K, Singh K, Patil P B, Oliva R, Patarapuwadol S, Bogdanove A J, Rai R. 2018. A strain of an emerging Indianpv.pathotype defeats the rice bacterial blight resistance genewithout inducing a clade III SWEET gene and is nearly identical to a recent Thai isolate., 9: 2703.
Chaijarasphong T, Thammachai T, Itsathitphaisarn O, Sritunyalucksana K, Suebsing R. 2019. Potential application of CRISPR-Cas12a fluorescence assay coupled with rapid nucleic acid amplification for detection of white spot syndrome virus in shrimp.,512: 734340.
Chen J S, Ma E B, Harrington L B, da Costa M, Tian X R, Palefsky J M, Doudna J A. 2018. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity.,360: 436‒439.
Chen P, Zhou J, Wan Y B, Liu H, Li Y Z, Liu Z X, Wang H J, Lei J, Zhao K, Zhang Y L, Wang Y, Zhang X H, Yin L. 2020. A Cas12a ortholog with stringent PAM recognition followed by low off-target editing rates for genome editing., 21(1): 78.
Creutzburg S C A, Swartjes T, van der Oost J. 2020. Medium- throughputdetection of DNA cleavage by CRISPR- Cas12a., 172: 27‒31.
Cui Z, Ojaghian M R, Tao Z, Kakar K U, Zeng J, Zhao W, Duan Y, Vera Cruz C M, Li B, Zhu B, Xie G. 2016. Multiplex PCR assay for simultaneous detection of six major bacterial pathogens of rice., 120(5): 1357‒1367.
Daher R K, Stewart G, Boissinot M, Bergeron M G. 2016. Recombinase polymerase amplification for diagnostic applications., 62(7): 947‒958.
Ding X, Yin K, Li Z Y, Lalla R V, Ballesteros E, Sfeir M M, Liu C C. 2020. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay., 11(1): 4711.
Garita-Cambronero J, Palacio-Bielsa A, López M M, Cubero J. 2017. Pan-genomic analysis permits differentiation of virulent and non-virulent strains ofthat cohabitspp. and elucidate bacterial virulence factors., 8: 573.
Gootenberg J S, Abudayyeh O O, Lee J W, Essletzbichler P, Dy A J, Joung J, Verdine V, Donghia N, Daringer N M, Freije C A, Myhrvold C, Bhattacharyya R P, Livny J, Regev A, Koonin E V, Hung D T, Sabeti P C, Collins J J, Zhang F. 2017. Nucleic acid detection with CRISPR-Cas13a/C2c2., 356: 438‒442.
Kanitchinda S, Srisala J, Suebsing R, Prachumwat A, Chaijarasphong T. 2020. CRISPR-Cas fluorescent cleavage assay coupled with recombinase polymerase amplification for sensitive and specific detection of., 27: e00485.
Lang J M, Hamilton J P, Diaz M G Q, van Sluys M A, Burgos M R G, Vera Cruz C M, Buell C R, Tisserat N A, Leach J E. 2010. Genomics-based diagnostic marker development forpv.andpv.., 94(3): 311‒319.
Lang J M, Langlois P, Nguyen M H R, Triplett L R, Purdie L, Holton T A, Djikeng A, Vera Cruz C M, Verdier V, Leach J E. 2014. Sensitive detection ofPathovarsandby loop-mediated isothermal amplification., 80(15): 4519‒4530.
Lau H Y, Botella J R. 2017. Advanced DNA-based point-of-care diagnostic methods for plant diseases detection., 8: 2016.
le Thanh T, Thumanu K, Wongkaew S, Boonkerd N, Teaumroong N, Phansak P, Buensanteai N. 2017. Salicylic acid-induced accumulation of biochemical components associated with resistance againstpv.in rice., 12(1): 108‒120.
Li S Y, Cheng Q X, Wang J M, Li X Y, Zhang Z L, Gao S, Cao R B, Zhao G P, Wang J. 2018a. CRISPR-Cas12a-assisted nucleic acid detection., 4: 20.
Li S Y, Cheng Q X, Liu J K, Nie X Q, Zhao G P, Wang J. 2018b. CRISPR-Cas12a has both- and-cleavage activities on single-stranded DNA., 28(4): 491‒493.
Liu H, Wang J B, Zeng H J, Liu X F, Jiang W, Wang Y, Ouyang W B, Tang X M. 2021. RPA-Cas12a-FS: A frontline nucleic acid rapid detection system for food safety based on CRISPR- Cas12a combined with recombinase polymerase amplification., 334: 127608.
Liu P P, Luk K, Shin M, Idrizi F, Kwok S, Roscoe B, Mintzer E, Suresh S, Morrison K, Frazão J B, Bolukbasi M F, Ponnienselvan K, Luban J, Zhu L J, Lawson N D, Wolfe S A. 2019. Enhanced Cas12a editing in mammalian cells and zebrafish., 47(8): 4169‒4180.
Lu W, Pan L Q, Zhao H J, Jia Y L, Wang Y L, Yu X P, Wang X Y. 2014. Molecular detection ofpv.,pv., andin infected rice seeds and leaves., 2(6): 398‒406.
Mahas A, Hassan N, Aman R, Marsic T, Wang Q C, Ali Z, Mahfouz M M. 2021. LAMP-coupled CRISPR-Cas12a module for rapid and sensitive detection of plant DNA viruses., 13(3): 466.
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. 2000. Loop-mediated isothermal amplification of DNA., 28: E63.
Piepenburg O, Williams C H, Stemple D L, Armes N A. 2006. DNA detection using recombination proteins., 4(7): e204.
Rudresha G V, Urs A P, Manjuprasanna V N, Suvilesh K N, Sharanappa P, Vishwanath B S. 2019. Plant DNases are potent therapeutic agents againstvenom-induced tissue necrosis in mice., 120(5): 8319‒8332.
Sullivan T J, Dhar A K, Cruz-Flores R, Bodnar A G. 2019. Rapid, CRISPR-based, field-deployable detection of white spot syndrome virus in shrimp., 9(1): 19702.
Wang X J, Ji P P, Fan H Y, Dang L, Wan W W, Liu S Y, Li Y H, Yu W X, Li X Y, Ma X D, Ma X, Zhao Q, Huang X X, Liao M. 2020. CRISPR/Cas12a technology combined with immune- chromatographic strips for portable detection of African swine fever virus., 3(1): 62.
Xiao G H, Zhang S, Liang Z H, Li G Q, Fang M T, Liu Y Y, Zhang J J, Ou M, He X, Zhang T Y, Zeng C C, Liu L, Zhang G L. 2020. Identification ofspecies and subspecies using the Cas12a/sgRNA-based nucleic acid detection platform., 39(3): 551‒558.
Xu H P, Zhang X L, Cai Z X, Dong X Q, Chen G, Li Z L, Qiu L M, He L, Liang B, Liu X L, Liu J F. 2020. An isothermal method for sensitive detection ofcomplex using clustered regularly interspaced short palindromic repeats/ Cas12aandcleavage., 22(8): 1020‒1029.
Xu L Q, Dai Q Q, Shi Z Y, Liu X T, Gao L, Wang Z Z, Zhu X Y, Li Z. 2020. Accurate MRSA identification through dual- functional aptamer and CRISPR-Cas12a assisted rolling circle amplification., 173: 105917.
Yasmin S, Hafeez F Y, Mirza M S, Rasul M, Arshad H M I, Zubair M, Iqbal M. 2017. Biocontrol of bacterial leaf blight of rice and profiling of secondary metabolites produced by rhizosphericBRp3., 8: 1895.
Yuan M, Ke Y G, Huang R Y, Ma L, Yang Z Y, Chu Z H, Xiao J H, Li X H, Wang S P. 2016. A host basal transcription factor is a key component for infection of rice by TALE-carrying bacteria., 5: e19605.
Zetsche B, Gootenberg J S, Abudayyeh O O, Slaymaker I M, Makarova K S, Essletzbichler P, Volz S E, Joung J, van der Oost J, Regev A, Koonin E V, Zhang F. 2015. Cpf1 is a single RNA- guided endonuclease of a class 2 CRISPR-Cas system., 163(3): 759‒771.
Zhang Y M, Zhang Y, Xie K. 2020. Evaluation of CRISPR/ Cas12a-based DNA detection for fast pathogen diagnosis and GMO test in rice., 40(1): 1‒12.
6 September 2021;
8 November 2021
Kittisak Buddhachat (kittisakbu@nu.ac.th); Kawee Sujipuli (kawees@nu.ac.th)
Copyright © 2022, China National Rice Research Institute. Hosting by Elsevier B V
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/)
Peer review under responsibility of China National Rice Research Institute
http://dx.doi.org/10.1016/j.rsci.2021.11.005
(Managing Editor: Wu Yawen)

- Rice Science的其它文章
- Polycomb Repressive Complex 2-Mediated H3K27 Trimethylation Is Required for Pathogenicity in Magnaporthe oryzae
- Genome-Wide Analysis of von Willebrand Factor A Gene Family in Rice for Its Role in Imparting Biotic Stress Resistance with Emphasis on Rice Blast Disease
- Selenium Alleviates Carbohydrate Metabolism and Nutrient Composition in Arsenic Stressed Rice Plants
- Feasibility of Improving Unmanned Aerial Vehicle-Based Seeding Efficiency by Using Rice Varieties with Low Seed Weight
- Genetic Variation for Anaerobic Germination and Emergence from Deeper Soil Depth in Oryza nivara Accessions
- Arsenic Accumulation in Rice:Sources, Human Health Impact and Probable Mitigation Approaches