
皮下脂膜炎样T细胞淋巴瘤临床分析
2022-06-16 02:34赵天娇魏一鸣李兆明
肿瘤基础与临床 2022年2期
阮 航, 张 蕾, 赵天娇, 魏一鸣, 李兆明
(郑州大学第一附属医院肿瘤科,河南 郑州 450052)
皮下脂膜炎样T细胞淋巴瘤(subcutaneous panniculitis-like T-cell lymphoma,SPTCL)是一种罕见的外周T细胞淋巴瘤亚型,主要累及皮下脂肪组织。SPTCL在整体人群中的发病率低于0.2/10万,主要临床表现为孤立或多发的皮下结节或红斑,好发于下肢及躯干,多数患者可出现发热、肝脾肿大等全身症状[1-2]。该病发展至后期可合并噬血细胞综合征(hemophagocytic lymphohistiocytosis,HLH),预后较差,需要临床医生及时诊断并治疗。由于发病率低,目前有关SPTCL的大样本临床分析有限,我们对10例SPTCL患者的临床资料进行回顾性分析,以提高人们对SPTCL的认识。
1 资料与方法
1.1 研究对象10例均为郑州大学第一附属医院病理科于2015年1月至2020年12月间确诊的SPTCL患者,病理诊断结果按WHO淋巴造血系统肿瘤分类。收集患者的临床及生物学资料,包括年龄、性别、临床症状、有无HLH、骨髓累及、病理结果、治疗方案及治疗反应。
1.2 治疗及随访10例患者均接受联合化疗,治疗结束后通过CT或PET-CT评价疗效,通过电话随访确认患者的生存情况。总生存期定义为从病理诊断之日起至因任何原因死亡或最后一次随访的时间。随访时间截止于2021年11月。通过电话随访询问患者目前的治疗和生存信息。
2 结果
2.1 一般临床特征10例SPTCL患者中,男5例,女5例,中位年龄为35.5(18~63)岁。其中年龄>60岁1例,年龄≤60岁9例,所有患者在确诊SPTCL之前均无自身免疫性疾病病史。7例最初表现为多发皮下结节和(或)斑块,2例表现为单发肿块,1例表现为蜂窝组织炎样皮肤损害。皮损受累部位以下肢(70%)和躯干(40%)多见,较少累及上肢(30%)及面部(20%)。除皮损外,1例(病例4)出现面部肿胀,1例(病例1)合并有右足水肿。5例就诊时出现B症状(包括发热、盗汗、体质量减轻),2例(病例1、3)复发时合并HLH。
影像学检查显示6例初诊时有淋巴结肿大,3例伴有肝脾肿大。7例行PET-CT检查,皮损处可见脂肪密度增高放射性分布浓聚,病变部位的最大SUV值3.2~9.2,其中1例(病例4)发现有肺部受累,1例(病例3)骨骼受累。1例(病例6)在初诊时提示骨髓浸润,病例3复发时淋巴瘤累及骨髓,骨髓液涂片可见吞噬细胞迹象。见表1。
2.2 病理及免疫组织化学结果10例均表现为皮下脂肪组织中非典型性淋巴细胞呈叶性或弥漫性脂膜炎样浸润,2例患者可观察到真皮层有轻微淋巴细胞浸润,未见表皮浸润。1例表现为皮肤肉芽肿性炎,伴大量淋巴组织增生。所有患者都可见淋巴样细胞倾向于包裹单个脂肪细胞形成花环样外观。浸润的淋巴细胞小至中等大小,细胞核大,形状不规则,核质深染。可见组织细胞浸润,脂肪坏死、核分裂相及核碎裂。未见小血管坏死或血管中心性浸润。
免疫组化显示肿瘤淋巴细胞除2例CD4+/CD8+表型外,其余均为CD4-/CD8+表型,CD3为阳性,缺乏CD20、CD56表达。7例患者进行βF1染色,6例结果为阳性。肿瘤细胞通常表达细胞毒颗粒相关蛋白,除病例4外,其余患者TIA-1染色均为阳性。8例进行颗粒酶B染色均为阳性。7例行TCR基因重排检测,其中6例存在单克隆性增生的T细胞群。10例EBER杂交均为阴性。Ki-67染色显示增殖指数介于30%~70%之间。见表2。
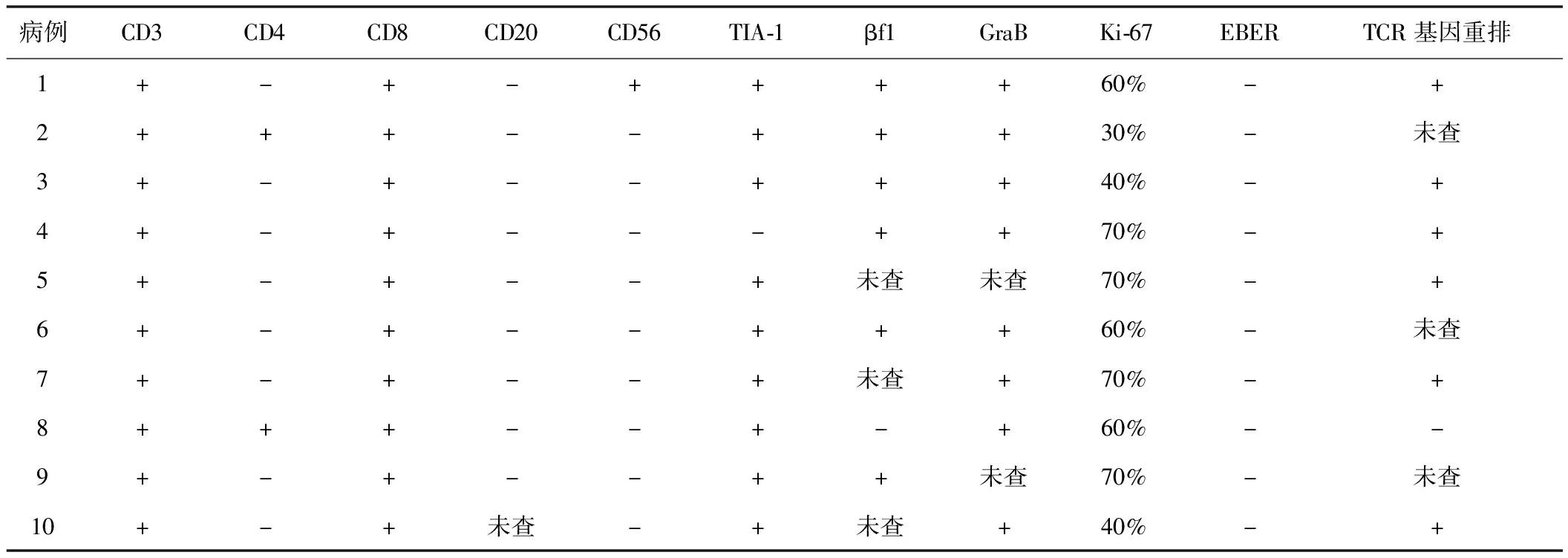
表2 10例SPTCL患者的免疫组织化学结果
2.3 治疗及预后所有患者均接受联合化疗作为初始治疗,完全缓解(CR)5例,部分缓解(PR)5例,总有效率为100%。5例患者接受吉西他滨、顺铂、地塞米松(GDP)方案治疗,3例获得长期CR,2例(病例1、3)疾病复发获得疾病进展(PD)合并HLH。4例患者接受地塞米松、顺铂、吉西他滨、培门冬酶(DDGP)方案治疗均获得长期缓解,病例6接受DDGP方案后更换为依托泊苷、顺铂、阿糖胞苷、地塞米松(ESHAP)方案,由PR达到CR,病例9接受DDGP方案化疗2个周期后出现肺栓塞,考虑到培门冬酶可导致血栓形成,后续更换环磷酰胺、阿霉素、长春新碱、地塞米松(CHOP)方案治疗。病例10一线接受阿霉素、泼尼松、阿糖胞苷、顺铂(ASHAP)方案达到PR,结束治疗4 a后病情复发并接受GDP方案挽救化疗达到CR。截至最后一次随访时,8例存活,2例死于SPTCL相关HLH,中位生存时间63.5(9~121)个月。8例幸存者无持续性皮损,2例患者(病例7、9)皮肤遗留局限性无症状的脂肪萎缩。见表1。
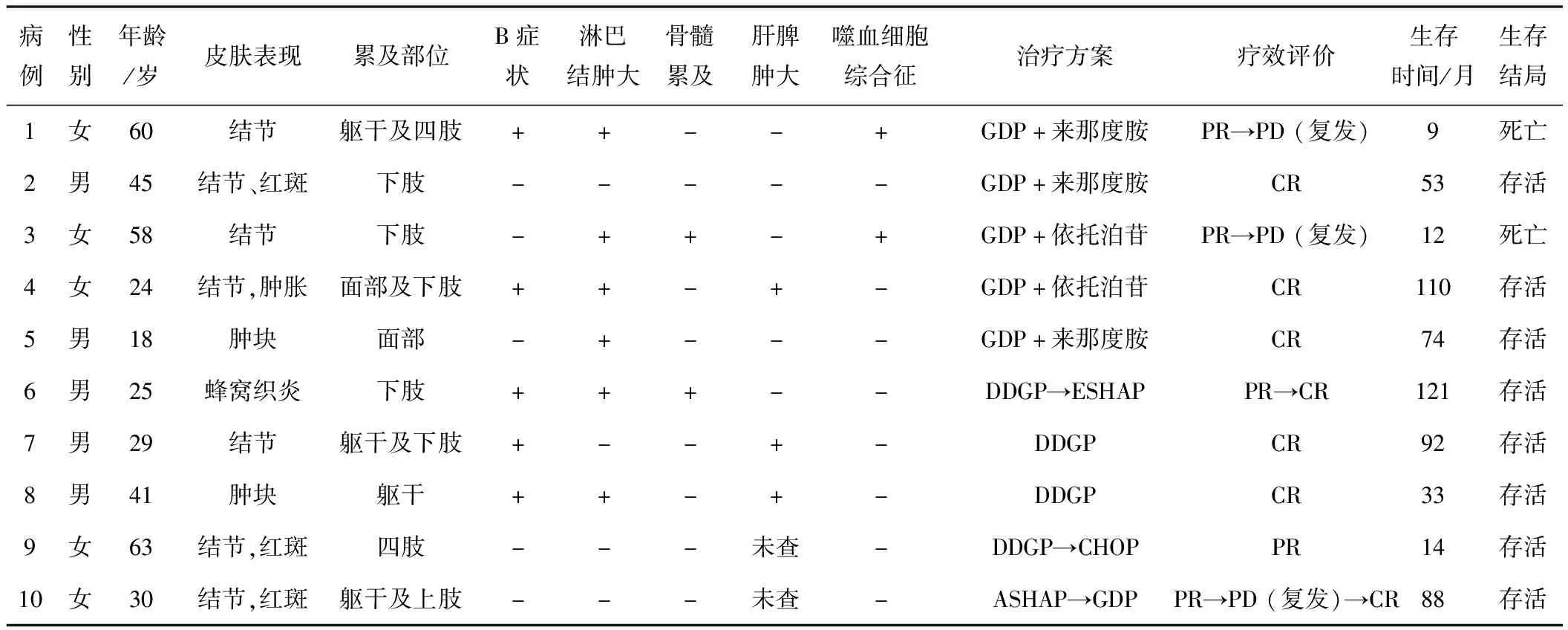
表1 10例SPTCL患者的临床病理特征及治疗转归
3 讨论
目前关于SPTCL的长期疗效研究很少,本研究总结了10例SPTCL患者的单中心经验,包括临床、病理和长期随访数据。
本研究中患者的临床表现与文献报道一致,皮损以孤立或多发的皮下结节为主,好发于下肢和躯干,1例表现为蜂窝组织炎样皮肤损害,需要与感染性疾病相鉴别,2例结节消退后遗留脂肪萎缩。多数患者出现B症状、淋巴结肿大及肝脾肿大等全身症状,骨髓累及少见。
SPTCL主要依靠病理活检确诊。除了CD4-/CD8+ SPTCL表型以外,本组还包含2例CD4+/CD8+表型。这2例患者化疗后生存时间为33~53个月,提示这种罕见表型与患者的临床病程无关。高Ki-67指数和TCR重排结果通常为排除自身免疫性疾病提供有价值的信息,本研究中所有患者Ki-67指数均大于30%,86%的患者存在TCR基因重排。过去普遍认为SPTCL的发生与EB病毒有一定相关性,在Kumar等[3]和Salhany等[4]研究的SPTCL组中,所有病例均未检测到EB病毒。这提示SPTCL的致病机制与EB病毒没有明确联系,本研究中所有病例EBER原位杂交均为阴性,进一步佐证了这一观点。因此对于EB病毒阳性的患者,需要警惕其他血液学肿瘤。一些观点认为,自身免疫性疾病患者发生SPTCL的风险更高,Willemze等[2]发表的SPTCL回顾性研究中,报告了19%的相关自身免疫性疾病病例。在Michonneau等[5]的研究中,40%的患者有自身免疫性疾病的病史,在65%的患者中检测到自身抗体。本组病例均无自身免疫性疾病病史,可能与样本量较小有关。
由于SPTCL发病率低,治疗方法因临床表现而异,缺乏大量数据和临床试验,目前尚未确定标准的治疗策略。既往化疗多采用基于蒽环类药物的方案,但缓解时间较短,且该类方案对复发难治患者效果差。在欧洲癌症研究与治疗组织2008年的研究中,非化疗组缓解率与化疗组相似,但非化疗组的复发率(56%)明显高于化疗组(10%)[2]。皮质醇或其他免疫抑制剂在SPTCL患者中作为单一疗法可提供短期的治疗反应,但一旦药物减少或停药,患者的病情会随之恶化。
GDP和DDGP是2种基于顺铂和吉西他滨的联合化疗方案。顺铂是一种金属基的化疗药物,具有广谱抗肿瘤活性,可与DNA上的嘌呤基相互作用,形成链内和链间交联,引起DNA损伤,从而发挥抗肿瘤作用[6]。吉西他滨是一种脱氧胞苷类似物,主要作用于S期,其与DNA聚合酶相互作用,通过模仿天然核苷酸的结构合并到DNA链中,从而干扰DNA的复制、转录或修复[7]。研究[8]表明,吉西他滨通过链间交联修复抑制机制与顺铂引起的DNA损伤具有协同抗肿瘤作用。此前,该类方案在其他淋巴瘤亚型中的疗效已有报道[9-11]。本研究中,这类以吉西他滨和顺铂为基础的化疗方案应用于SPTCL同样效果显著。
SPTCL合并HLH死亡率高达63%~80%,预后较差[2]。文献中目前尚无关于SPTCL合并HLH的最佳治疗方案的描述。Alaibac等[12]认为对于具有侵袭性病程的SPTCL患者,更积极的方案,包括高剂量化疗(high-dose treatment,HDT)和干细胞移植(stem cell transplantation,SCT)是一种合适的选择。Medhi等将[13]BFM-90方案用于伴有HLH的SPTCL患者,获得了长期缓解。后续也有研究[14-15]报道了BFM-90方案在SPTCL和HLH的治疗效果,进一步支持大剂量化疗方案对继发性HLH的价值。HDT-SCT在少数个案报道中被证实有效,在台湾的报告中,3例合并HLH的患者接受了造血干细胞移植,均获得完全缓解,持续时间超过30个月[16]。对于疾病快速进展的患者,可选择HDT-SCT。环孢素是一种钙调神经磷酸酶抑制剂,可以下调细胞因子的表达,零星报道证实在合并HLH的患者中,单用环孢素或与皮质醇联合使用可以获得良好的效果[17]。然而,需要更多的临床研究来验证这一点。
SPTCL作为一种罕见疾病,并发HLH的患者预后较差,确诊应结合临床表现和病理检查。SPTCL目前尚无推荐的治疗指南,对于初治或复发SPTCL患者,以顺铂和吉西他滨为基础的化疗方案是一种合适的治疗选择。未来还需要更大样本量的前瞻性研究来阐明此类方案在治疗SPTCL中的作用。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
健康护理(2022年3期)2022-05-26
中国典型病例大全(2022年12期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
现代临床医学(2022年1期)2022-02-12
现代仪器与医疗(2021年6期)2022-01-18
特别健康·下半月(2019年9期)2019-09-24
文萃报·周二版(2019年26期)2019-09-10
时代英语·高二(2017年4期)2017-08-11
第二课堂(课外活动版)(2015年4期)2015-10-21
