
spICP-MS准确定量多分散金纳米颗粒的颗粒数量浓度
2022-06-15 04:02:26李秀城巢静波董丽洁房叶天董硕飞
质谱学报 2022年3期
李秀城,巢静波,董丽洁,房叶天,2,董硕飞
(1.中国计量科学研究院化学所,北京 100029;2.北京化工大学,北京 100029;3.安捷伦科技(中国)有限公司,北京 100102)
金纳米颗粒(AuNPs)作为一种常见的金属纳米材料,因具有化学性质稳定、制备方法成熟、易于表面修饰等特点,被广泛应用于生物监测[1-3]、癌症治疗[4-6]、药物递送[7-9]等领域。纳米颗粒在生产和使用过程中很容易进入环境,有研究表明,AuNPs能够积累在生物体内,并对多种器官造成损伤[10],这引发了人们对其潜在风险的关注。纳米颗粒毒性与颗粒数量浓度密切相关,其作为评价纳米颗粒样品的重要参数,直接决定纳米颗粒样品的性质。因此,对纳米材料的定量不仅应针对粒径和质量浓度,还应考察颗粒数量浓度。
常见的颗粒数量浓度测定方法包括理论计算、纳米颗粒追踪分析(NTA)等。理论计算法根据测得的颗粒质量浓度和粒径计算颗粒数量浓度,采用透射电子显微镜(TEM)测定粒径,但测得的粒径难以反映样本的总体情况,而且离子的存在会使质量浓度的测定结果存在较大偏差。NTA法基于光散射原理,易受空气和样品杂质干扰,且颗粒数量浓度检出限较高,难以对环境样品中痕量的纳米颗粒进行定量。此外,在实际的环境样品中,由于纳米颗粒易发生团聚、溶解等变化,有可能以含有多种粒径颗粒的多分散形式存在。然而,当前环境中纳米颗粒的定量研究主要针对只含有单一粒径分布的单分散样品[11-13],故亟需开发一种针对多分散纳米样品的分析方法。常见的多分散测定方法主要包括场流分级(FFF)[14]、水动力学色谱(HDC)[15]、尺寸排阻色谱(SEC)[16]和毛细管电泳(CE)[17]等,但大多存在尺寸分辨率差、回收率低,且只能给出样品的质量浓度等缺点,难以得到颗粒数量浓度信息。
单颗粒电感耦合等离子体质谱(single particle inductively coupled plasma mass spectrometry,spICP-MS)检出限低(低至ng/L级),基体干扰小,可快速获取纳米颗粒的粒径分布、质量浓度和颗粒数量浓度等信息,被广泛应用于纳米材料的定量和表征。2021年,Chao等[18]采用spICP-MS测定了单分散AuNPs的颗粒数量浓度,测定结果与配制值一致,证明了该技术的测量准确性。此外,有文献证明了spICP-MS定量分析环境样品[19]、生物样品[20]和消费品[13]中纳米颗粒的可靠性。鉴于spICP-MS在测量纳米颗粒中的优势,有研究尝试使用spICP-MS对多分散颗粒分散液进行定量。Liu等[21]使用spICP-MS对颗粒数量浓度比为1∶1的30 nm和60 nm多分散AuNPs进行表征,发现能够完全区分颗粒信号。Donahue等[22]使用spICP-MS对颗粒数量浓度比为1∶1的30 nm与55 nm AuNPs混合样品进行分析,发现不同粒径的颗粒信号峰区分明显,且各组分颗粒通量与混合比例一致,但并没有测定颗粒数量浓度,也没有对影响多分散定量的条件进行探究。
本研究拟采用spICP-MS法测定包含30 nm和60 nm AuNPs的多分散样品的颗粒数量浓度,考察载气流速、采样深度、采集时间、驻留时间对颗粒数量浓度测定结果的影响。在优化的条件下,对不同混合比例的多分散样品进行定量测定,以验证该方法对不同多分散样品的适用性。采用建立的方法测量自来水、泉水和湖水中AuNPs的颗粒数量浓度,并对3种水样进行加标回收实验,以验证方法的实用性,旨为实际环境中多分散纳米颗粒的准确定量提供技术支持。
1 实验部分
1.1 主要仪器与装置
8900型电感耦合等离子体质谱仪:美国Agilent公司产品,配有单颗粒分析模块和内径1.0 mm的炬管;JEM-2100F型透射电子显微镜:日本JEOL公司产品;NanoSight NS300 纳米颗粒跟踪分析仪:英国Malvern公司产品;XP204型电子分析天平:瑞士Mettle-Toledo 公司产品;超纯水机:美国Millipore公司产品;iCAP 7400 DUO型电感耦合等离子体发射光谱仪:美国Thermo Fisher公司产品。
1.2 主要材料与试剂
柠檬酸钠(纯度99%):美国Alfa Aesar公司产品;AuNPs分散液(粒径30 nm和60 nm):英国BBI公司产品;金溶液标准物质(GBW08650,质量浓度100 μg/g):中国计量科学研究院产品,使用时用1 mmol/L柠檬酸钠溶液稀释至1 μg/kg;AuNPs质控样品(LGCQC5050,颗粒数量浓度1.47×1011NPs/g,质量浓度45.1 mg/kg):英国政府化学家实验室产品,使用时用1 mmol/L柠檬酸钠溶液稀释至20 ng/kg;调谐液(Li、Co、Y、Ce、Tl质量浓度均为1.0 μg/L):美国Agilent公司产品;自来水取自实验室,泉水取自北京市凤凰岭,湖水取自北京市圆明园,所有水样在上机前过0.22 μm滤膜。
1.3 实验条件
1.3.1仪器条件 射频功率1 550 W;雾化器:Micromist nebulizer;监测同位素:197Au;数据采集模式:TRA;分析模式:no gas;等离子气流速15.0 L/min;载气流速0.6~1.0 L/min;采样深度7.0~10.0 mm;采集时间60~240 s;驻留时间:0.1、0.2、0.5、1.0、3.0 ms;蠕动泵转速0.1 r/s;雾化室温度2 ℃;氧化物比率<1.5%,双电荷比率<2%。
1.3.2测定原理 在单颗粒模式下,AuNPs依次进入等离子体中电离形成离子云,检测器检测到离子云后产生颗粒信号,信号数量与颗粒数量浓度成正比,信号强度与颗粒质量成正比。Pace等[23]提出式(1)用于计算纳米颗粒的颗粒数量浓度。
(1)
其中,NNP为纳米颗粒的颗粒数量浓度,f(INP)为检测到的颗粒信号数量,qliq为样品提升速率(g/min),ti为采集时间(min),ηn为传输效率。
传输效率使用纳米颗粒标准物质或质控样品按照式(2)进行测定。
(2)
其中,NRM为标准物质或质控样品的颗粒数量浓度,f(IRM)为检测到的标准物质或质控样品的颗粒信号数量。
1.3.3实验过程 室温下,在50 mL聚丙烯离心管中加入适量的30 nm和60 nm AuNPs,按重量法用1 mmol/L柠檬酸钠溶液稀释。为避免在单位驻留时间内出现2个及以上的纳米颗粒,将配制样品的颗粒通量控制在800~1 200 NPs/min。样品稀释完成后超声30 s,防止纳米颗粒团聚。分析前使用1.0 μg/L的Li、Co、Y、Ce、Tl调谐液对仪器进行调谐,使m/z7、89、205的信号灵敏度最高,且氧化物、双电荷比率分别小于1.5%和2%。将进样管插入1 mmol/L 柠檬酸钠溶液中持续吸取溶液5 min,记录质量变化,计算提升速率。测定过程中,以1.0 μg/kg金离子溶液计算响应因子,以20 ng/kg LGCQC5050纳米颗粒分散液计算样品的传输效率。为避免残留在管路中的AuNPs对测定结果造成干扰,样品之间依次以3%(V/V)稀王水、3%(V/V)重蒸硝酸和Milli-Q超纯水清洗管路。测量完成后,通过软件的选区积分功能对多分散样品中不同粒径的AuNPs进行定量分析。
2 结果与讨论
2.1 单分散AuNPs颗粒数量浓度的测定
有研究表明,spICP-MS在3 ms驻留时间下能够准确测定单分散样品的粒径和颗粒数量浓度[18,24]。基于此,本实验首先在3 ms驻留时间下分别测定30 nm和60 nm AuNPs两种单分散样品的粒径和颗粒数量浓度,并将测定结果与TEM测得的粒径值和NTA测得的颗粒数量浓度值进行比较,结果列于表1。可见,spICP-MS测定值的重复性较好,且与TEM和NTA的测定结果一致。因此,分别以1.34×1014NPs/kg和1.98×1013NPs/kg作为30 nm和60 nm AuNPs的颗粒数量浓度值。

表1 spICP-MS与TEM、NTA测定结果的比较(n=7)Table 1 Comparison of spICP-MS with TEM and NTA measurement results (n=7)
2.2 实验条件优化
2.2.1尺寸分辨率的优化 当使用spICP-MS分析多分散AuNPs时,为保证测量结果的准确性,颗粒的尺寸分辨率需足够高。由于纳米颗粒的信号强度与其粒径的3次方成正比,可以通过提高信号响应改善尺寸分辨率。载气流速和采样深度(采样锥锥孔到负载线圈之间的距离)是决定信号响应的2个重要参数,当载气流速过小或采样深度过大时,离子在等离子体中的扩散时间延长,使进入采样锥的离子减少,造成信号响应下降;当载气流速过大或采样深度过小时,样品在等离子体中的时间过短,发生不完全电离,使电离产生的离子减少,同样造成信号响应的下降[25]。2017年,Kálomista等[26]研究了采样深度对金、银纳米颗粒信号值的影响,结果显示,采样深度能够显著影响颗粒的信号响应和尺寸分辨率,在最佳的信号响应条件下,尺寸分辨率最高。2019年,Kinnunen等[25]研究了等离子体射频功率、载气流速和采样深度对金纳米颗粒和金离子信号响应的影响,结果表明,金纳米颗粒与金离子的信号强度呈相似的变化趋势,且优化后的信号响应提高了70%。本实验以1.0 μg/kg金离子标准溶液(Au+)和20 ng/kg的30 nm AuNPs分散液为研究对象,研究载气流速和采样深度对离子和颗粒信号响应的影响,示于图1。以颗粒数量浓度比1∶1的多分散样品(30 nm和60 nm AuNPs的颗粒数量浓度均为5.0×107NPs/kg)为研究对象,研究载气流速和采样深度对颗粒尺寸分辨率的影响,示于图2。参考Kálomista等[26]提出的分辨率公式,采用30 nm和60 nm AuNPs颗粒信号峰峰底距离d作为分辨率评价标准,d越高,分辨率越好,示于式(3)。
d=S60 nm-S30 nm-RW30 nm-LW60 nm
(3)
其中,d为30 nm和60 nm AuNPs颗粒信号峰峰底的距离,S60nm和S30nm分别为60 nm和30 nm AuNPs信号峰最高点对应的信号强度,RW30 nm为30 nm AuNPs信号峰的右峰宽,LW60 nm为60 nm AuNPs信号峰的左峰宽。
实验发现,Au+与AuNPs的信号强度具有相似的变化趋势,在采样深度不变时,载气流速对信号响应有显著影响。选取载气流速0.8 L/min时,Au+和AuNPs的信号响应最高,约为1.0 L/min时的3倍,且该流速下的尺寸分辨率最高。在0.8 L/min的载气流速下,观察采样深度对信号响应的影响,发现相比于载气流速,采样深度对仪器的信号响应影响相对较小,这与Kinnunen等[25]的结论一致,但与Kálomista等[26]的发现不同,可能是因为Kálomista等在实验中采用了稀释气和较长的驻留时间(6 ms)。当采样深度为9 mm时,Au+和AuNPs的信号响应最大,且在该条件下,30 nm和60 nm信号的分离度最好。因此,选择载气流速0.8 L/min,采样深度9 mm。
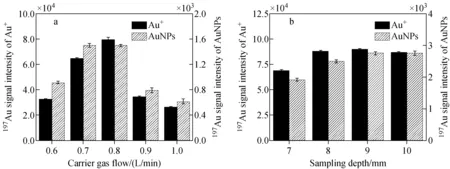
注:a.采样深度8 mm,采集时间60 s,驻留时间3 ms;b.载气流速0.8 L/min,采集时间60 s,驻留时间3 ms图1 载气流速(a)和采样深度(b)对仪器信号响应的影响Fig.1 Effect of carrier gas flow rate (a) and sampling depth (b) on instrument sensitivity
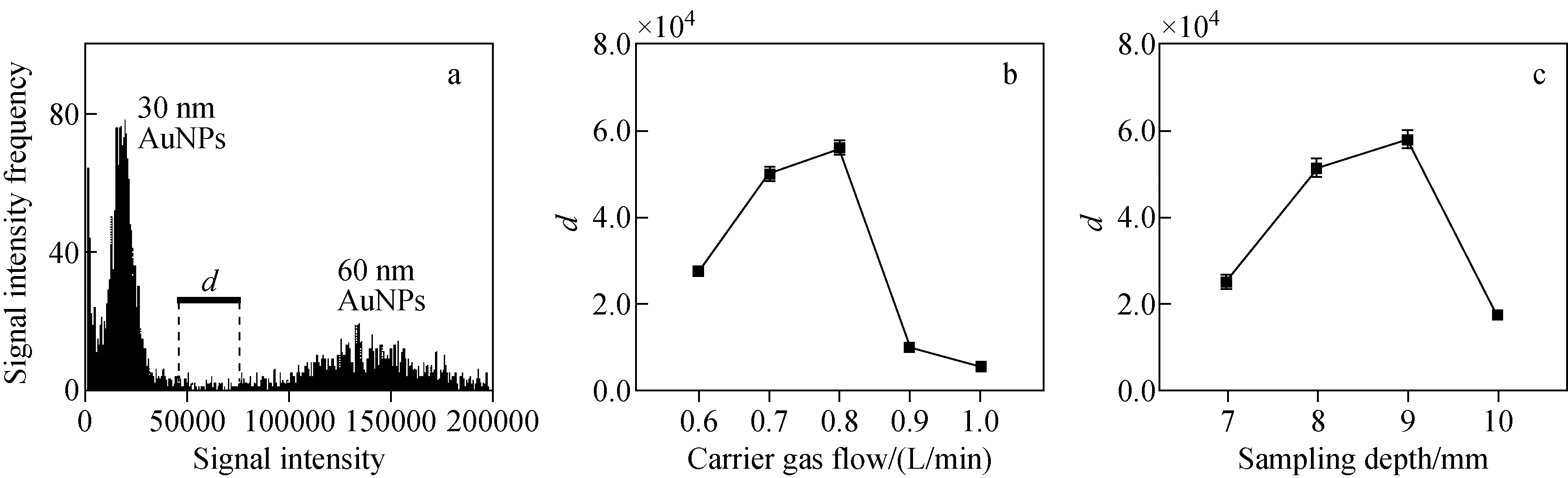
注:a.分辨率示意图;b.采样深度8 mm,采集时间60 s,驻留时间3 ms;c.载气流速0.8 L/min,采集时间60 s,驻留时间3 ms图2 载气流速(b)和采样深度(c)对尺寸分辨率的影响Fig.2 Effect of carrier gas flow rate (b) and sampling depth (c) on size resolution
2.2.2采集时间的优化 以spICP-MS测定低浓度颗粒分散液时,较低的颗粒通量使测定结果的相对标准偏差(RSD)变大,为了准确定量低浓度颗粒分散液,需要降低RSD。根据Laborda等[27]提出的公式(4),颗粒数量浓度RSD的2次方与采集时间成反比,因此在通量不变时,延长采集时间可降低测定结果的RSD,提高结果的精密度。本实验依次选择60、90、120、150、180、210、240 s作为样品采集时间,对颗粒数量浓度比10∶1的AuNPs多分散样品(30 nm和60 nm颗粒数量浓度分别为7.0×1011NPs/kg和7.0×1010NPs/kg)进行定量,示于图3。不同采集时间下的测定结果基本一致,且随着采集时间的延长,60 nm颗粒数量浓度测定值的RSD逐渐下降,当采集时间增加至180 s时,RSD降至5%以下,能够满足分析要求。然而,过长的采集时间会增加分析时长,为了在准确定量的前提下缩短分析时间,选择180 s作为多分散样品的采集时间。
(4)

注:载气流速0.8 L/min,采样深度9 mm,驻留时间3 ms图3 采集时间对颗粒数量浓度结果的影响Fig.3 Effect of acquisition time on the results of particle number concentration
2.2.3驻留时间的优化 驻留时间(dwell time,tdwell)是影响spICP-MS测定值的重要参数之一[28],其能够直接决定分析结果的质量。当驻留时间过长时,容易发生多颗粒事件;而驻留时间过短时,因单次采集的信号值过低,由随机误差带来的影响将会相当显著。本实验采用0.1、0.2、0.5、1.0、3.0 ms驻留时间对颗粒数量浓度比10∶1的30 nm和60 nm AuNPs多分散样品进行分析,结果示于图4。由于1个纳米颗粒的信号持续时间在0.3~1.0 ms之间,因此在0.1、0.2、0.5、1.0 ms驻留时间下会产生瞬态信号(由颗粒某一部分电离生成的信号),需要采用“峰积分模式”(将多个瞬态信号求和以得到单个纳米颗粒信号强度的模式[28]);而在3.0 ms驻留时间下,由于驻留时间长于信号持续时间,因此无需采用“峰积分模式”。3.0 ms和0.1 ms驻留时间的信号分布示于图5。结果显示,随着驻留时间的缩短,检测结果出现偏差,这可能是由于在较短的驻留时间下会出现“颗粒分割事件”(将1个颗粒的信号误认为是2个颗粒的信号);当驻留时间降至0.1 ms时,测得值与配制值相当,这是因为此时颗粒峰的分辨率最高,能够有效避免“颗粒分割事件”的发生,从而避免了可能出现的偏差。此外,较高的分辨率有利于小粒径颗粒与离子背景的区分,30 nm颗粒信号受离子的干扰更小,示于图6,故选择0.1 ms作为驻留时间。
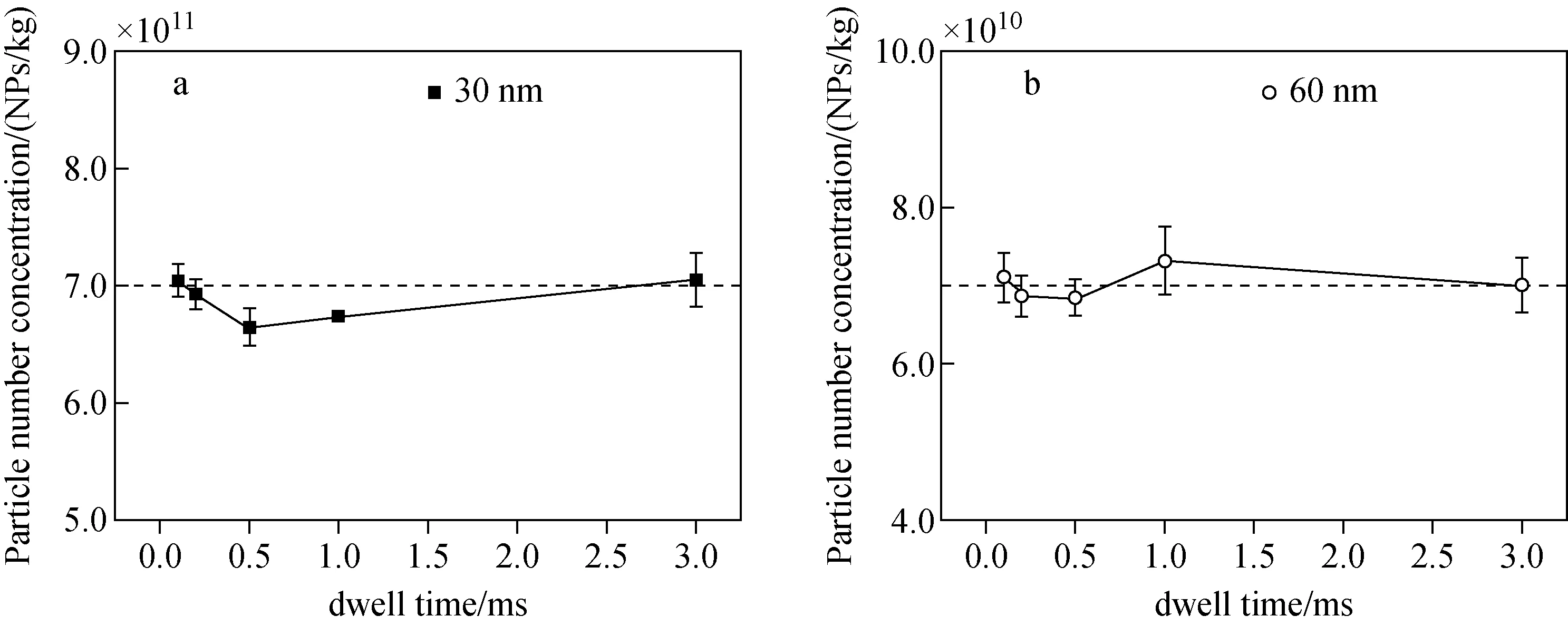
注:载气流速0.8 L/min,采样深度9 mm,采集时间180 s图4 驻留时间对颗粒数量浓度结果的影响Fig.4 Effect of dwell time on the result of particle number concentration
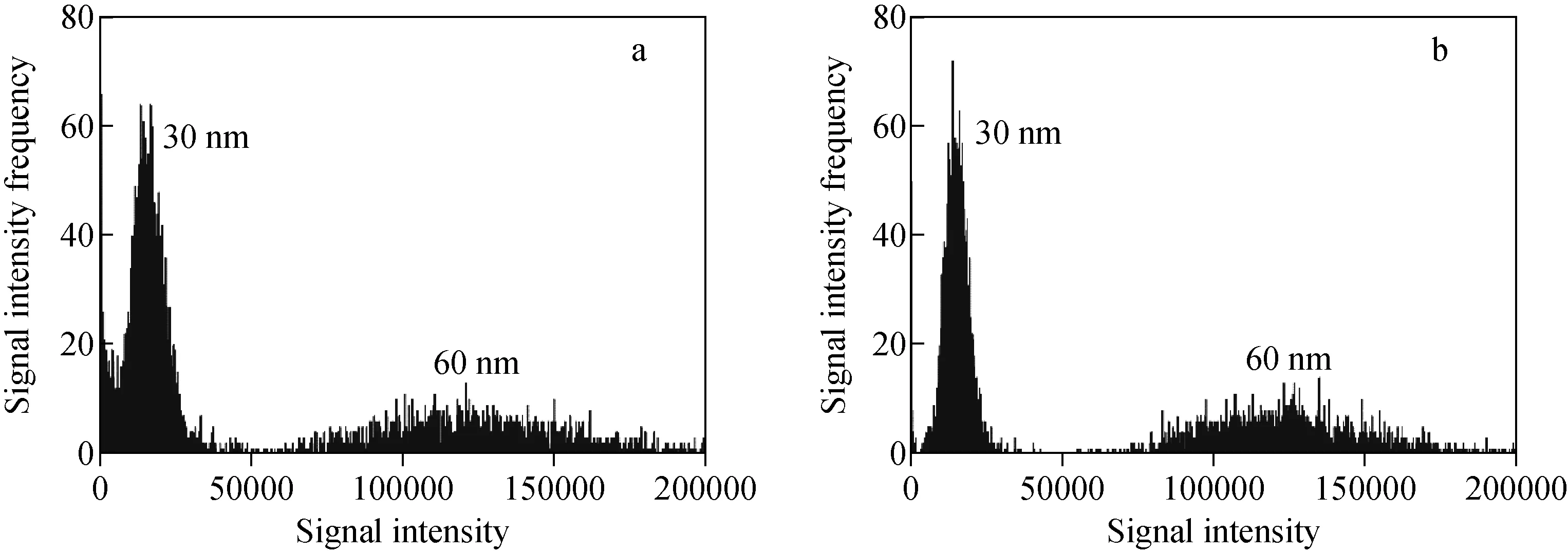
注:载气流速0.8 L/min,采样深度9 mm,采集时间180 s图5 驻留时间为3 ms(a)和0.1 ms(b)时的信号分布Fig.5 Signal distribution at dwell time of 3 ms (a) and 0.1 ms (b)
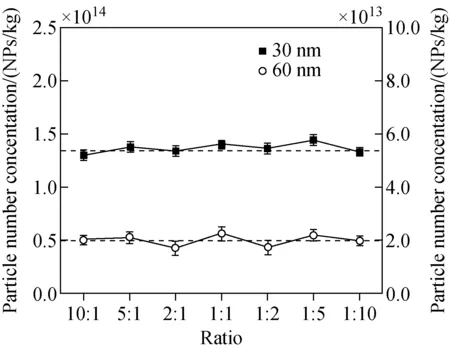
图6 不同混合比例多分散样品颗粒数量浓度测定结果Fig.6 Results of the particle number concentration of polydisperse samples with different mixing ratios
2.3 方法性能参数
2.3.1方法的准确性和重复性 本实验配制了30 nm和60 nm AuNPs颗粒数量浓度比10∶1的多分散AuNPs,在优化的条件下重复测定7次颗粒数量浓度值,结果列于表2。可见,30 nm和60 nm的颗粒数量浓度均与配制的浓度值相符,且RSD小于5%,证实了该方法的可靠性。
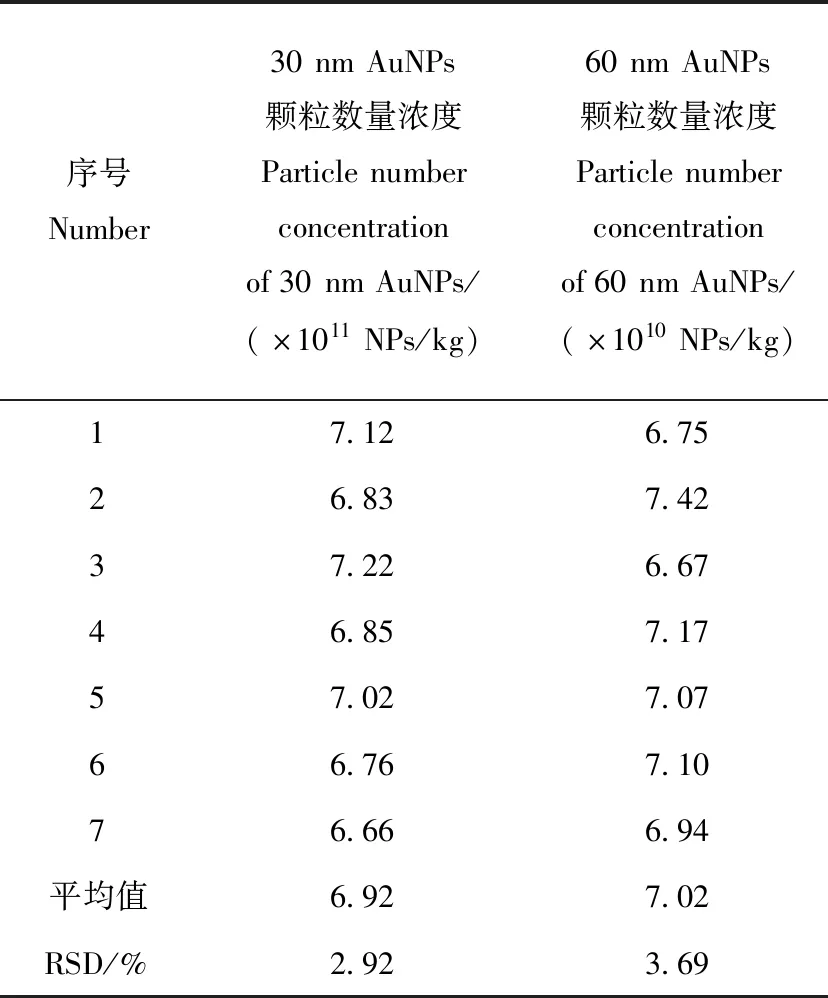
表2 重复性测定结果(n=7)Table 2 Results of repeatability (n=7)
2.3.2方法的适用性 使用已知颗粒数量浓度的30 nm和60 nm AuNPs分别配制不同颗粒数量浓度比(10∶1、5∶1、2∶1、1∶1、1∶2、1∶5、1∶10)的AuNPs分散液,并在优化的条件下对其颗粒数量浓度进行定量,分析完成后将测定结果与单分散的30 nm和60 nm AuNPs的颗粒数量浓度进行比较,示于图6。结果表明,在不同的混合比例下,测得结果与30 nm和60 nm AuNPs的颗粒数量浓度基本一致,说明该方法能够较好地分析不同混合比例的多分散样品。
2.3.3方法的检出限 根据式(5)和(6)可得到方法的粒径检出限LODsize和颗粒数量浓度检出限LODNP[27,29],经计算,分别为10 nm和45 NPs/g。
(5)
(6)
其中,σ为空白信号的标准偏差(cps),ηi为“按尺寸计算”的传输效率,qliq为样品提升速率(g/min),tdwell为驻留时间(ms),K为通过离子标准计算的响应因子(cps/(μg/kg)),fa为目标元素在纳米颗粒中的质量分数,ρ为纳米颗粒密度(g/cm3),ηn为“按浓度计算”的传输效率,ti为采集时间(min)。
2.4 实际环境水样中多分散AuNPs的测定
采用已优化的方法分别对自来水、泉水和湖水中的多分散AuNPs进行定量,3种水样中均未检出AuNPs。以低、中、高3个加标水平分别向自来水、泉水和湖水中加入30 nm和60 nm AuNPs,并计算加标回收率,结果示于图7。可见,在各加标浓度下,30 nm和60 nm AuNPs在自来水、泉水、湖水中的加标回收率均处于80%~120%之间,表明该方法适用于实际环境水样中多分散AuNPs的测定。对于30 nm AuNPs,在各加标浓度下,自来水和泉水中的加标回收率均高于湖水;对于60 nm AuNPs,在各加标浓度下,自来水中的加标回收率均高于泉水和湖水。综上可知,对于加标30 nm和60 nm AuNPs,湖水基体中的加标回收率最低,这可能是由于湖水中的钾、钠、钙、镁含量最高,列于表3,在一定程度上促进了纳米颗粒的团聚[30],从而导致回收率测定结果偏低。

注:低、中、高水平的加标浓度分别为1.0×107,2.0×107,5.0×107 NPs/kg图7 自然水体中AuNPs的加标回收率测定(n=3)Fig.7 Recovery rate of AuNPs in natural water (n=3)
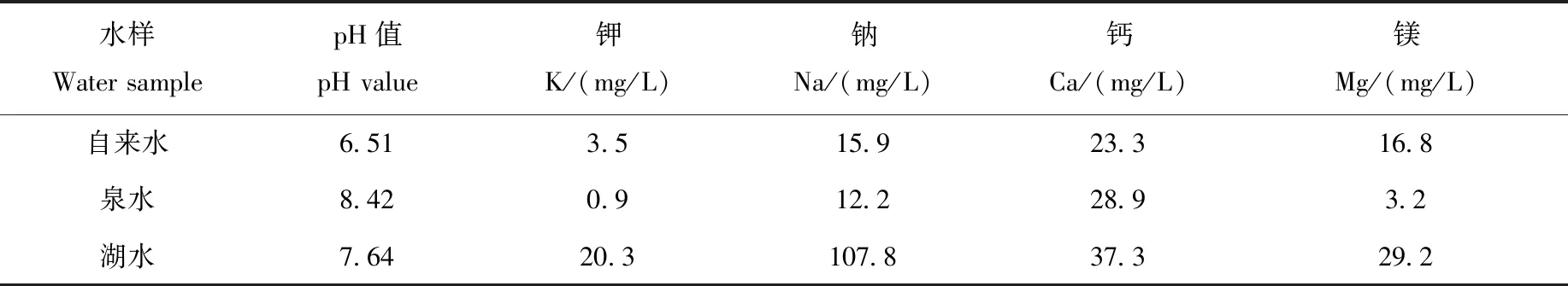
表3 水样成分的测定结果(n=3)Table 3 Measurement results of water sample composition (n=3)
3 结论
本研究建立了一种基于单颗粒电感耦合等离子体质谱的多分散AuNPs颗粒数量浓度的定量方法,以含有30 nm和60 nm AuNPs的多分散样品为研究对象,探究载气流速、采样深度、采集时间和驻留时间对分析结果的影响。在载气流速0.8 L/min,采样深度9 mm时,能以最佳的分辨率区分30 nm和60 nm AuNPs。在此基础上,采用采集时间180 s和驻留时间0.1 ms,实现了以不同比例混合的样品中30 nm和60 nm AuNPs颗粒数量浓度的准确定量,2种粒径AuNPs颗粒数量浓度测定结果的相对标准偏差小于5%。相比于此前的多分散研究,本研究在准确定量多分散体系中极低浓度颗粒组分方面具有较大优势,有望应用于实际环境水样中痕量金属纳米颗粒的迁移转化研究。
猜你喜欢
当代化工研究(2023年18期)2023-10-19 09:45:58
中学生数理化·八年级物理人教版(2023年4期)2023-05-05 07:29:42
中学生数理化·八年级物理人教版(2022年4期)2022-04-26 14:11:16
中国食用菌(2021年10期)2021-11-04 06:23:20
大众科学(2020年7期)2020-10-26 09:24:30
甘肃科技(2020年21期)2020-04-13 00:33:32
小天使·六年级语数英综合(2018年1期)2018-10-08 09:32:50
科学与财富(2018年22期)2018-08-18 11:06:32
北京航空航天大学学报(2017年2期)2017-11-24 05:24:51
科技资讯(2016年4期)2016-06-11 06:26:56
