
棉花籽指数量性状位点(QTL)的初步定位
2022-06-09 06:58李蓓姚金波李燕朱守鸿房圣涛陈伟张永山
中国棉花 2022年3期
李蓓,姚金波,李燕,朱守鸿,房圣涛,陈伟,张永山
(中国农业科学院棉花研究所/ 棉花生物学国家重点实验室,河南 安阳 455000)
棉花为我国纺织业提供了重要原材料,棉花种子作为重要的食用油原料,同样具有重要的经济价值[1]。 棉花的籽指影响籽棉产量、衣分、种子储存性能、出苗率及棉花苗期的生长势等性状[2-4]。 定位和克隆棉花籽指相关的基因,能为改良棉花籽指等性状提供基因资源,为开展棉花产量相关调控机制的研究和分子育种奠定基础。
棉花籽指与种子大小、种子质量和短绒数量等性状相关。 已有报道显示,在不同群体中得到的与棉花籽指关联的数量性状位点 (quantitative trait loci,QTL)往往位于多个不同染色体或区间,而且单个位点的遗传效应往往较低[5-6],表明籽指受多基因调控,属于典型的数量性状。
传统的QTL 定位具有周期长、 工作量大等缺点。 随着测序技术的不断发展,基于第2 代高通量测序与混合群体分离分析(bulked segregant analysis,BSA)的BSA-seq 技术可对双亲和混池进行高通量测序,而无须对整个定位群体的所有单株进行基因型分析,具有成本低、效率高的特点,适合质量性状位点或QTL 的初步定位[7]。BSA-seq 技术已经被广泛应用于水稻[8]、玉米[9]、大豆[10]等作物的QTL 定位研究,并且已在棉花中有一定的应用[11],但目前国内还未见利用BSA-seq 对籽指QTL 定位的报道。
1 材料与方法
1.1 供试材料
以陶小铃为母本、 大桃棉为父本进行杂交,收获F1种子。 其中:陶小铃来源于海陆杂交后代,植株整体偏小,叶面积及棉铃较小,果枝较长;大桃棉的叶面积、铃均较大,但植株紧凑,果枝较短。 F1自交后收获F2种子。2020 年4 月份将亲本及F2种植于中国农业科学院棉花研究所东场试验基地(河南省安阳县)。
1.2 试验方法
1.2.1样本采集。 开花期采集亲本(40 个单株)及F2群体(1 149 个单株)的嫩叶,置于2 mL 离心管中,-80 ℃保存。 采用改良的十六烷基三甲基溴化铵 (cetyl trimethyl ammonium bromide, CTAB)法提取DNA,并测定DNA 的质量浓度,-20 ℃保存备用。
1.2.2籽指调查。 成熟期分单株采摘中部棉铃10个,晾晒后轧花,每株随机挑选100 粒饱满种子称量。
1.2.3BSA 混池测序。 在F2群体中挑选具有极端籽指表型的植株, 分别构建 “大籽指”(含32 个单株)混池和“小籽指”混池(含28 个单株)。将每个池内单株DNA 样品等量混合, 将混池DNA 以及亲本的DNA 样品送深圳华大基因公司进行基因组重测序。 测序平台为BGISEQ-500。 亲本测序深度为20×,混池测序深度为30×。去除测序原始数据的接头[测序读长(reads)中接头序列长度超过读长全长50%以上则删除整条读长],过滤未知碱基(N)比例超过10%的读长, 去除超过50%碱基的质量值低于10 的读长,得到质控后的数据(clean reads)。
1.2.4QTL 定位分析。 采用BWA(Burrow-Wheeler Aligner Tool) 软件将质控后的测序数据比对到TM-1 参考基因组 (ZJU 2.1 版本)[12]。 采用GATK(The Genome Analysis Toolkit) 软件进行单核苷酸多态性(single nucleotide polymorphism, SNP)与插入缺失突变(insertion-delete mutation, InDel mutation)检测[13]。 筛选亲本间纯合多态性位点,采用基于SNP-index(亲本陶小铃某位点上含有SNP 的读长数与混池中该位点总读长数的比值)的QTL-seq 定位分析方法[14]。在2 个混池内分别计算SNP-index,用小籽指池的SNP-index 减去大籽指池的SNPindex 计算得到每个位点的Δ(SNP-index),然后分别计算每个位点95%和99%水平的Δ(SNP-index)置信区间,作为QTL 的筛选阈值。
2 结果与分析
2.1 亲本及F2 代群体籽指特征
亲本之间籽指差异明显, 父本籽指均值为14.7 g,母本籽指均值为7.8 g。 F2群体籽指表现为超亲分离,分布在5.81~19.11 g(图1)。 正态性检验表明,F2群体籽指符合正态分布(平均值为11.44 g,偏度为0.082 6,峰度为0.247 9,P=0.269 2),呈现典型的多基因控制的数量性状遗传特点。
2.2 混池测序分析
如图1,选取F2群体中籽指分布两端的单株构建混池。 其中,“大籽指” 混池籽指范围为15.10~19.11 g,“小籽指”混池籽指范围为5.81~7.90 g。对上述2 个混池和2 个亲本的DNA 进行第2 代高通量测序分析,获得质控后的数据。 全基因组中测序覆盖度≥20×的区段占比均超过90%,Q20 均大于96%(表1), 说明4 个文库的测序结果满足BSA-seq 分析要求。

图1 F2 群体棉花籽指的分布
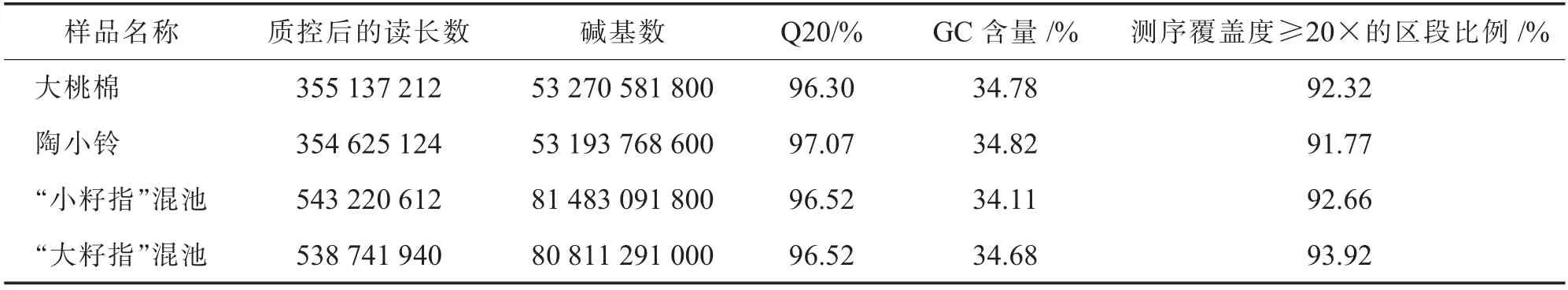
表1 测序质量和基因组覆盖度统计
2.3 QTL 定位分析
在2 个亲本间共筛选出纯合有差异的SNP 和InDel 多态性位点3 861 792 个, 通过BSA-seq 方法,绘制了多态性位点的Δ(SNP-index)在染色体上的分布图(图2)。Δ(SNP-index)值越高,表示2个极端混池在该区间的SNP-index 差异越大,该区间与籽指的相关性越强。 在A07、A13、D10 染色体上,共检测到3 个在0.05 水平显著的区间,其具体信息见表2。 其中,A07 染色体显著区段最长,跨度达49 Mbp,其Δ(SNP-index)值最高的区段(峰值区间)为60.3~61.3 Mbp,峰值区间内包含2 个基因,但均未注释,功能未知;A13 染色体显著区段的峰值区间为3.3~4.3 Mbp, 峰值区间内包含81 个基因;D10 染色体显著区段的峰值区间为7.2~8.2 Mbp,峰值区间内包含59 个基因。
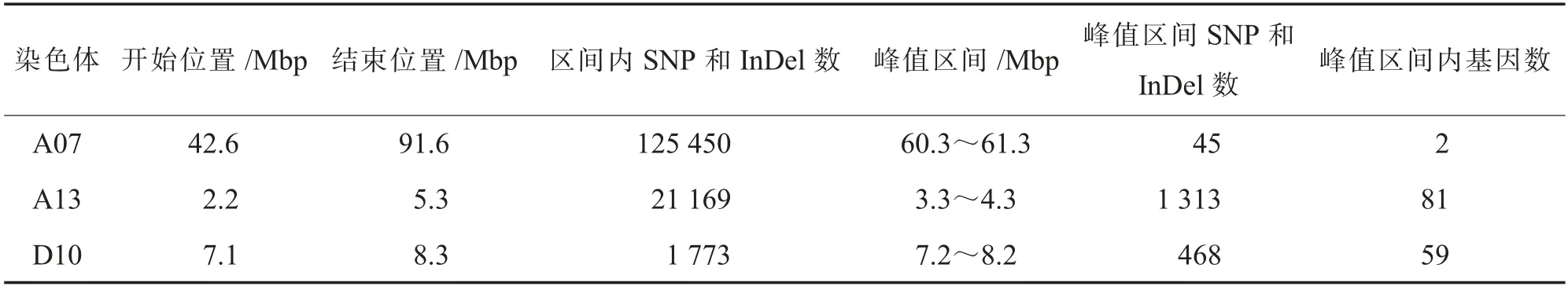
表2 候选区间信息
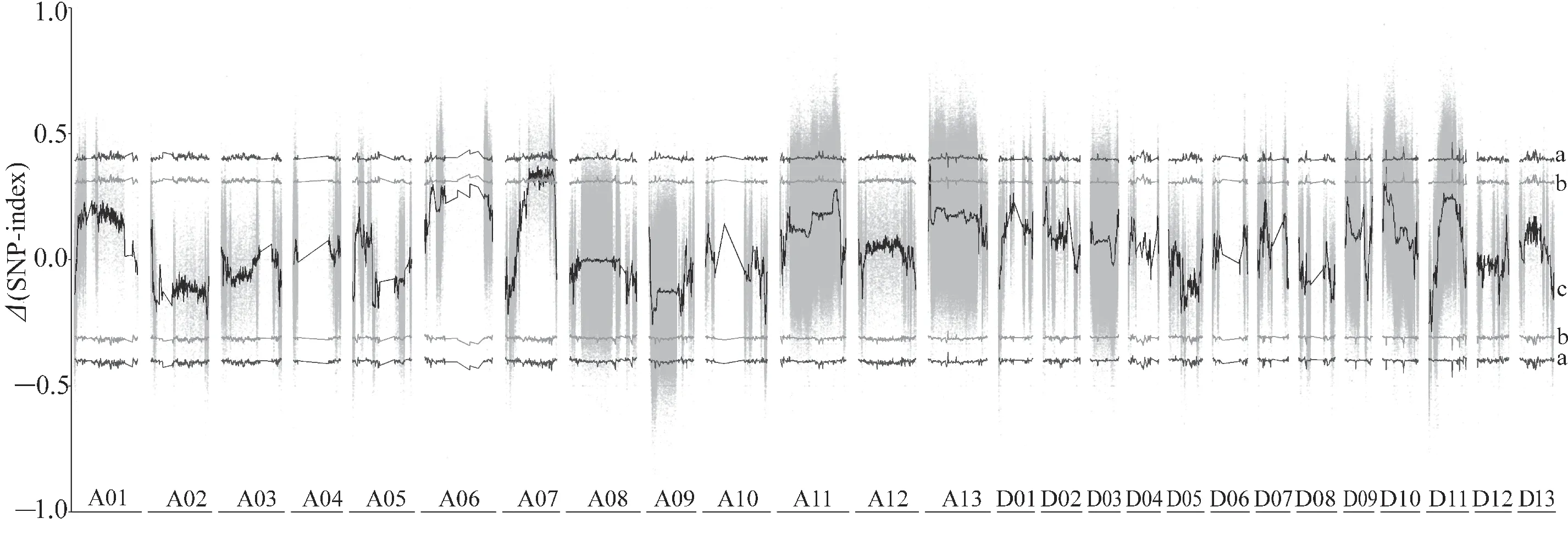
图2 多态性位点Δ(SNP-index)的分布
3 讨论与结论
棉花种子含有丰富的蛋白质[15],同时还是重要的油料来源[16],具有重要的经济价值[17]。 棉花籽指受多个相关性状的影响,如种子大小、质量、短绒密度等。 籽指是重要的产量决定因子之一[18],与衣分呈显著负相关[19-20];同时,也对棉花的生长发育具有重要的影响,如出苗率、苗期生物量等[4]。 目前对棉花籽指相关的基础研究相对薄弱, 已经成为了制约棉花育种创新的瓶颈, 因此加强棉花籽指的研究刻不容缓[21]。
本研究选择遗传背景差异较大的亲本大桃棉和陶小铃作为籽指的研究对象, 调查发现其F2群体单株的籽指符合正态分布, 属于典型的数量性状,且表现出超亲现象。 采用BSA-seq 技术分别在A07、A13、D10 染色体上均初步定位到1 个与籽指相关的QTL。 目前候选的定位区间共3 个,长度约为1.2~49.0 Mbp, 仍需QTL 精细定位以筛选有效的候选基因。 QTL 定位精度取决于控制该性状的基因数目以及基因与其附近标记的交换频率,而BSA-seq 由于仅利用了混池单株的基因型信息,一般存在精度较低的缺陷[14]。 但是,前人报道表明,BSA-seq 技术定位的峰值区间往往包含目的基因[22]。本研究定位的3 个峰值区间共包含142 个基因,这些基因可作为重点候选基因用于后续研究。
刘大军等[23]利用陆地棉重组近交系群体鉴定了9 个与籽指相关的QTL,分别定位于A02、A06、A09、A11、A12、D01、D02、D05 和D12 染 色 体 上;伊海法[24]利用陆地棉重组近交系群体也检测到3个与籽指相关的QTL,分别位于A03、D07、D09 染色体上。前人定位结果与本研究定位结果没有重合的区间,究其原因,可能有2 个方面:首先,由于籽指是1 个复合性状, 受到多个遗传因素的控制,不同研究使用的材料间存在不同的位点分离,关联位点重合度较低;其次,本研究使用的小籽指材料来源于海陆杂交后代,遗传背景与普通陆地棉差异较大,故定位的QTL 可能为前人未报道的新位点。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
西南农业学报(2022年5期)2022-06-06
安徽农学通报(2022年6期)2022-04-07
林业科技(2020年2期)2020-10-14
农民致富之友(2020年27期)2020-09-12
三农资讯半月报(2020年13期)2020-07-31
农业科技与装备(2018年1期)2018-12-10
电脑知识与技术(2018年19期)2018-11-01
