
氮掺杂生物质碳材料催化剂的制备及其催化CH4-CO2重整性能
2022-06-01 01:55秦晓伟张晓娣张国杰
洁净煤技术 2022年5期
马 晓,秦晓伟,张晓娣,张国杰
(1.洛阳瑞泽石化工程有限公司,河南 洛阳 471003;2.太原理工大学 省部共建煤基能源清洁高效利用国家重点实验室,山西 太原 030024)
0 引 言
近年来由于温室气体排放不断增加,气候变暖成为全球性问题。CO2作为主要的温室气体之一,其排放问题一直是关注焦点[1-2]。通过CH4-CO2重整反应,不仅可以实现CH4和CO2的有效转化,而且产生H2/CO≤1的合成气,适用于F-T合成的原料(如甲醇和二甲醚)[3],具有较好的经济价值。因此,CH4-CO2催化重整研究在石油、天然气化工以及合成化学领域关注较多[4]。在重整反应中,高效催化剂的开发是关键。目前重整催化剂的开发主要集中在贵金属和过渡金属催化剂两大类[5]。其中,贵金属催化剂在重整反应中表现出较高的催化活性和抗积碳性。但从经济角度来看,贵金属由于价格高昂限制了其工业化应用。而镍基非贵金属催化剂因其低成本和可媲美贵金属的催化活性被广泛研究。然而,镍基催化剂在重整反应过程中易积碳和烧结,从而导致催化剂失活[6]。因此,要实现CH4-CO2重整反应制合成气工业化,迫切任务之一就是寻找廉价且具有良好性能的催化剂。
碳基材料由于价格低廉,且具有较大的比表面积、较强的吸附能力和丰富的表面官能团等特性,作为非再生型非金属催化剂引起了广泛关注[7]。澳大利亚西澳大学张东柯院士课题组[8]考察了非再生型煤焦碳材料对CH4-CO2重整反应的作用,证实了煤焦炭材料中的碳结构在共转化过程中对甲烷转化具有催化作用。国内东南大学肖睿课题组[9]将不同煤种制得的碳材料直接作为重整反应的非再生型催化剂,发现不同煤种制得的碳材料在重整过程中具有不同的催化活性,其中具有较高比表面积和较低灰分的碳材料催化活性相对较高;CH4和CO2在不同碳材料上的转化行为具有相同趋势,初始转化率较高,然后逐渐降低并趋于稳定。FIDALGO等[10]将具有不同孔隙结构和表面含氧官能团的活性炭用作重整反应的非再生型催化剂,发现活性炭的孔隙结构和表面含氧官能团都对其催化性能具有重要影响。XU等[11]进一步考察了HNO3和NaNO3改性对AC碳材料催化重整性能的影响,发现改性前后不同碳材料催化剂在低温下的催化活性有很大差异,在高温下差异不明显;同时发现改性碳材料中的羟基在CH4-CO2重整中起重要作用。所以,通过调控碳材料的孔结构和表面化学性质可以提升非再生型非金属碳基材料催化剂的催化性能。
通过文献调研发现,掺杂杂原子(N、B、S、P等)可以改善碳材料的物理化学性能[12]。其中,由于N原子的半径和结构性质与C原子非常接近,因此被广泛用作杂原子掺杂到碳材料中。在碳材料中引入N官能团,不仅可以增加催化剂表面的碱性位点,还可以促进反应过程中的电子转移,从而提高N掺杂碳材料的催化性能[12]。GONG等[13]将制备的N掺杂碳纳米管直接用作氧化还原反应的电催化剂时,展现出比商用Pt/C催化剂更好的催化活性和稳定性。LONG等[14]通过试验证明了N掺杂碳材料中的C—N 基团是催化ORR反应的活性中心;而且发现C—N基团的种类和数量与制备过程条件有直接关系。ZHOU等[15]研究发现,在碳材料中引入N原子,能有效增强其对原料气的吸附能力,从而提高材料的催化活性;但认为掺杂形成的石墨型N是催化剂的活性中心。虽然试验已经证明掺杂引入的N原子是催化反应的活性中心,但目前仍无法证明某种特定的N形式与反应活性之间的准确关系,其反应机理也存在争议,有待进一步深入研究。
生物质碳材料因可再生、价格低、来源广等优点成为碳材料领域的研究热点。其中,农业废弃物的木屑中含有丰富的纤维素、半纤维素和木质素,是制备多孔碳材料的理想原料[16-19]。为加深N掺杂碳材料非再生型催化剂对CH4-CO2重整催化作用内在规律的认识,笔者以木屑为碳源、尿素为N源、KOH为活化剂,采用不同的N掺杂方式制备了一系列N掺杂生物质碳材料非再生型催化剂,并将其用于CH4-CO2重整反应。采用比表面孔结构分析仪、电镜、红外、和热重等表征对新鲜和使用后的非再生型催化剂样品的结构和特性进行分析,探究不同N掺杂方式对制备N掺杂生物质碳材料的比表面积、孔结构和N官能团等的影响。并对反应前后非再生型催化剂样品的结构和特性进行分析,探究N掺杂生物质碳材料的孔结构和氮官能团对重整反应的影响。
1 试 验
1.1 试剂
试验所用的碳源木屑购自惠丰秸秆农产品深加工公司;分析纯级化学试剂尿素和KOH购自天津科密欧化学试剂有限公司;CH4、CO2、N2、H2和Ar等气体购自太原钢铁集团比欧西气体有限公司,纯度为99.99%。
1.2 催化剂样品制备
首先,将木屑和尿素按质量比1∶1进行物理混合;然后将混合均匀的样品置于加热炉中,在350 ℃N2气氛下碳化2 h,升温速率为5 ℃/min;碳化得到的样品被标记为NBC。将KOH和NBC按质量比3.5∶1.0进行物理混合并研磨,然后将混合样品与前述碳化在同样条件下,750 ℃活化2 h,得到的样品洗涤以除去多余KOH;将样品洗涤到中性后干燥12 h,得到样品被标记为NBC-750/3.5(原位掺N)。仅木屑在上述条件350 ℃碳化获得的样品标记为BC;进一步,采用KOH在750 ℃活化得到的样品标记为BC-750/3.5。将BC-750/3.5样品和尿素按照质量比1∶1进行物理混合,然后在350 ℃ N2气氛下碳化2 h,获得的样品记为BC-750/3.5-N(后处理掺N)。
1.3 样品活性测试
将2.5 g样品置于固定床反应器中进行活性测试,石英管反应器直径为60 mm。反应前,首先通入5 min N2,排出反应器及样品中吸附的空气等杂质。原料气CH4和CO2总流量为120 mL/min,体积比为1∶1。原料气的流量由质量流量计控制,温度由程序控温仪控制,升温速率为7.5 ℃/min。尾气经干燥处理后,采用在线气相色谱分析其气体成分。CH4和CO2的转化率为
X(CH4)=(FinC(CH4)in-Fout×
C(CH4)out)/(FinC(CH4)in)×100%,
(1)
X(CO2)=(FinC(CO2)in-Fout×
C(CO2)out)/(FinC(CO2)in)×100%,
(2)
式中,X(CH4)为CH4转化率,%;X(CO2)为CO2转化率,%;Fin为入口气总流量,mL/min;Fout为出口气总流量,mL/min;C(CH4)in为原料气中CH4体积分数,%;C(CH4)out为出口气CH4体积分数,%;C(CO2)in为原料气中CO2体积分数,%;C(CO2)out为出口气中CO2体积分数,%。
1.4 样品表征
样品的孔结构特性采用贝士德表面分析仪(3H-2000PS2型)获得;测试前,样品在250 ℃脱气4 h。根据获得的吸附-脱附等温线,按照BET多点法获得样品的总比表面积;按照BJH法获得孔体积和孔径分布;按照DFT法获得样品的微孔孔径;按照N吸附数据获得孔径分布曲线。催化剂样品的形貌结构通过蔡司扫描电镜(MERLIN Compact型)获得。催化剂样品的元素组成通过ELEMENTAR vario EL III型元素分析仪获得。样品的官能团通过红外光谱仪(默飞IS5型)获得。样品中的N物种类型由X射线光电子能谱仪(Thermo ESCALAB 250XI 型)获得。样品中的碱性位采用化学吸附(AutoChem1 II 2920型)获得。样品反应后热稳定性曲线采用德国耐驰(STA449F3型)热重分析仪获得。
2 结果与讨论
2.1 不同催化剂的孔结构特性分析
表1为新鲜及反应后不同催化剂样品的孔结构特性参数。由表1可知,新鲜样品BC的比表面积和孔体积分别为11.25 m2/g和0.03 mL/g;经过原位掺杂N后样品NBC的比表面积和孔体积降低到4.57 m2/g和0.02 mL/g。表明原位氮掺杂改变了材料的孔道结构和表面性质。而经KOH活化后的样品BC-750/3.5的比表面积(1 314.59 m2/g)和孔体积(0.71 mL/g)明显提高。但后处理N掺杂后,得到的BC-750/3.5-N样品的比表面积和孔体积降低,孔径增大。这主要是由于,一方面在引入N过程中破坏了BC-750/3.5的孔结构;另一方面由于引入的部分N源没有完全分解而留在孔道内及其表面,堵塞了部分孔道。与其他材料相比,原位掺杂活化制备的NBC-750/3.5样品具有最大的比表面积(1 981.53 m2/g)和孔体积(1.08 mL/g),平均孔径也较小(2.18 nm)。这说明原位N掺杂后,掺杂N的碳材料在活化时由于N的协同作用,更有利于KOH刻蚀造孔,从而形成较好的孔结构。大比表面积和孔体积有利于反应原料气与活性位点接触,从而提高材料的催化活性。重整反应后,样品BC和NBC比表面积均增加,这主要是由于二者的碳化温度较低(350 ℃),高温(900 ℃)重整反应过程中部分具有较强活性的碳及其含氧官能团等被消耗,从而产生新孔。BC-750/3.5-N样品反应后比表面积和孔体积增加也是同样的原因。
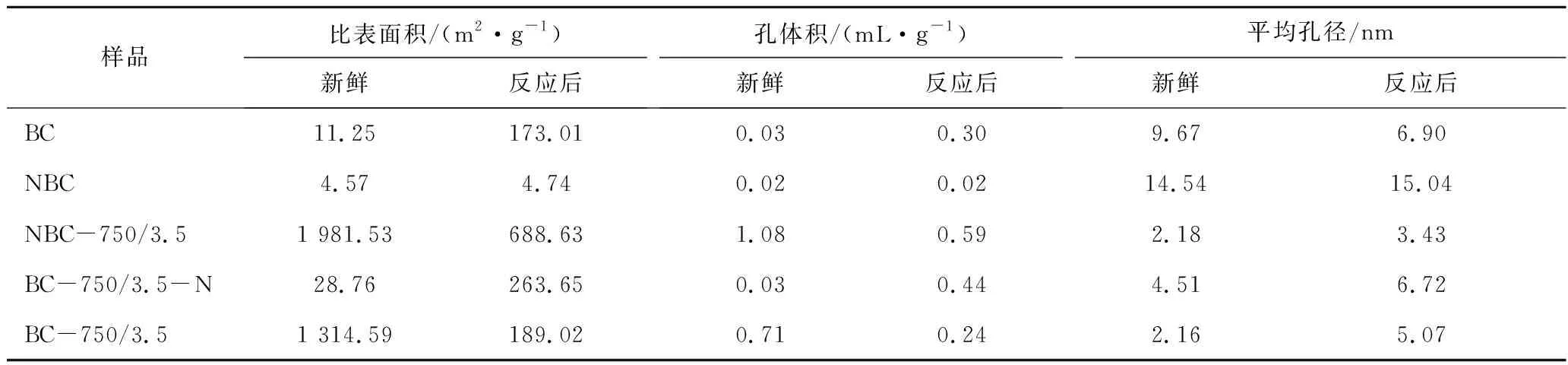
表1 新鲜和反应后催化剂的孔结构参数
图1为不同样品新鲜及反应后样品的吸-脱附等温线和孔径分布。由图1(a)和(d)可知,反应前后BC和BC-750/3.5-N样品的吸脱附等温线相同。反应前由于二者比表面积和孔容较低,因此其吸脱附等温线都较低;反应后二者的吸脱附等温线在低压(P/P0)下呈Ⅰ型等温线,在高压下呈Ⅳ型等温线,且带有H3型滞后环,这说明样品含有微孔和介孔[20-21],这与图1(f)的孔径分布一致。由图1(b)可知,新鲜和重整反应后NBC样品的吸脱附等温线基本相同,而且没有形成闭合;由图1(f)和(g)可知,含有少量介孔。由图1(c)和(e)可知,反应前后NBC-750/3.5和BC-750/3.5样品的吸脱附等温线相同。二者的新鲜样品吸脱附等温线呈Ⅰ型,说明二者的新鲜样品主要由微孔组成,这也与图1(f)的孔径分布相一致。样品中含有的微孔有利于CO2吸附,从而增加与活性位点的接触,提升样品的催化性能。反应后二者的吸脱附等温线在低压(P/P0)下呈I型等温线,在高压下呈Ⅳ型等温线,且带有H3型滞后环,说明高温重整反应使材料孔结构发生变化。对比图1(f)和(g)可知,重整反应后,仅原位掺杂制备的NBC-750/3.5 样品保留了较好的孔径结构,因此显示出最佳的催化活性和稳定性。由上述分析可知,不同N掺杂方式对所制备催化剂样品的孔结构和催化剂样品的催化性能具有重要影响。
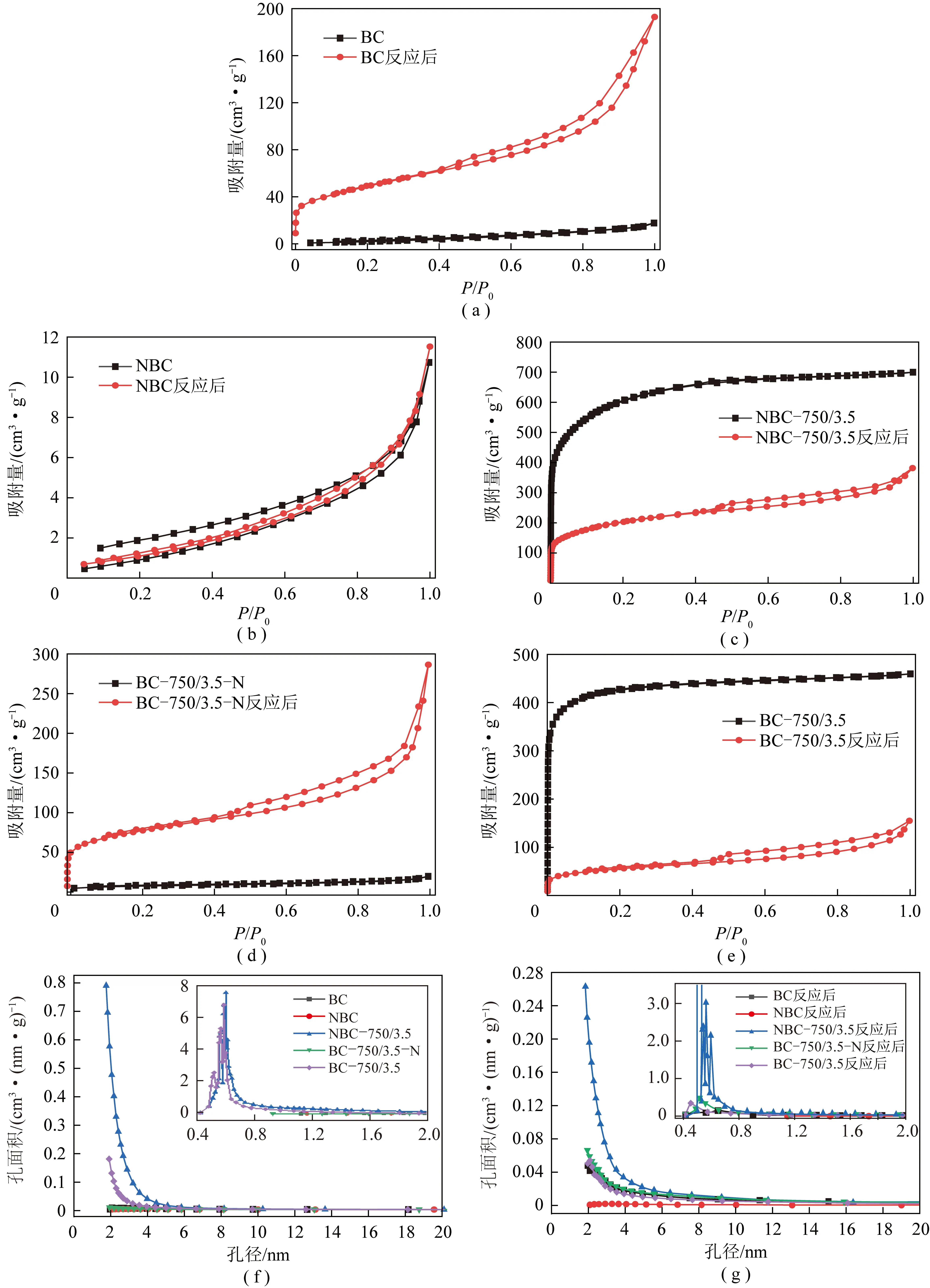
图1 反应前后不同催化剂的N2吸附脱附等温线和孔径分布曲线Fig.1 Nitrogen adsorption and desorption isotherms and the pore size distribution curves of different catalysts fresh and spent
2.2 样品的表面形态结构
图2为不同样品的表面形态结构。可知仅碳化的BC样品表面十分光滑,没有明显的孔隙结构,因此其比表面积和孔容较低,与前述孔结构特性分析结果一致,催化效果也较差。在添加尿素共碳化的NBC样品,表面出现1层类灰物质,因此其比表面积和孔容降低;NBC样品经过进一步活化后,得到的NBC-750/3.5样品呈多孔状,因此其比表面积和孔容也明显增加,这有利于样品对反应物分子CH4和CO2的吸附和活化[22]。由图2(d)可知,原位掺杂的NBC-750/3.5样品孔径结构主要由随机分布的蠕虫状微孔组成,从而提高了反应气体分子与活性位点的接触,降低了过程传质阻力,提高了材料的催化性能[23]。
2.3 催化剂元素分析
不同样品的元素分析结果见表2。可知,新鲜样品BC中O含量较高,质量分数达20.10%;反应后,样品O质量分数降至2.21%。这表明,在重整反应过程中含O官能团被消耗。结合反应前后材料的性能可知,材料中所含的官能团是催化剂活性的重要组成部分[24-25]。从表2中还可以发现,与尿素共碳化后,催化剂样品NBC的N质量分数从原来的0.13%提高到10.49%;进一步活化后,得到的NBC-750/3.5样品的N质量分数明显降低(1.96%),但仍高于仅碳化的BC样品,这说明N被成功引入到材料中。而活化后样品的N含量降低,一方面是由于表面引入的N不稳定,高温发生了分解或重排;另一方面是由于活化过程中发生了KOH刻蚀,降低了C和N含量。与原位N掺杂制备的NBC-750/3.5 相比,活化后掺杂N得到的BC-750/3.5-N样品的N质量分数明显提高,高达26.35%。较高的N质量分数有利于提高催化剂稳定性,因此重整过程中材料的稳定性保持较好。与反应前的生物质碳相比,反应后所有样品N和O质量分数都明显降低,表明重整反应过程中含O官能团和含N官能团被消耗,因此反应后其催化活性降低。另外,对比可知,并不是所制备的碳材料中N含量越高,其催化活性就越高,这说明所制备碳材料的活性不仅由掺杂的N含量决定。结合前述材料的孔结构特性分析可知,所制备碳材料的性能主要是由其孔结构和N官能团类型和含量等共同决定。
2.4 样品红外表征
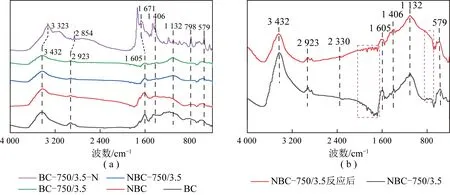
图3 不同催化剂样品红外光谱Fig.3 FT-IR spectroscopy of different catalysts
2.5 样品的XPS表征分析
图4为不同样品的XPS N1s谱图,表3为根据图4的XPS N1s分峰拟合的数据。根据文献[27]可知,398.3 eV位置处的峰为吡啶型N(N-6)的特征峰,400.1 eV 位置处的峰为吡咯型N(N-5)的特征峰,401.5 eV 位置处的峰为石墨型N(N-Q)的特征峰。由图4和表3可知,在制备的所有样品中,BC样品中的吡啶N占比最高,达71%,这有助于提高样品BC催化活性,因此其在初始时表现出较高的催化活性。N-6和N-5在NBC样品质量分数基本相同,分别占48%和51%。活化后BC-750/3.5样品中的N-6从原来的71%降至29%,减少的N-6少量转化为N-5,大部分被转化为N-Q。表明通过改变活化条件和制备方式,可以控制碳材料生成N的种类和含量,从而实现催化剂性能的可控制备。与BC样品不同的是,NBC样品活化后所得NBC-750/3.5样品中的N-5质量分数与NBC样品相比无变化,部分N-6转化为N-Q。根据文献[28-29]可知,吡咯氮N-5对CO2具有较好的吸附作用,吡啶氮N-6碱性较强,在二者的共同作用下提高了所制备材料对CO2的吸附和活化能力,从而提高了所制备碳材料的催化活性。因此,具有较高N-6和N-5的NBC-750/3.5样品具有较好的重整催化活性和稳定性。反应后NBC-750/3.5样品的N-6和N-5质量分数分别由原来的31%和50%分别降至5%和4%,N-Q占比从原来的19% 提高到91%。表明重整反应时,不仅部分具有活性的含N官能团(N-6和N-5)被消耗,还有部分在反应中发生了转变,生成性质稳定的N-Q,从而使催化剂活性先降低后稳定。由表3可知,BC-750/3.5-N样品中几乎不存在N-Q,这是由于后处理掺N反应温度较低(350 ℃),因此掺杂N的种类主要以N-6和N-5的形式存在。上述分析表明,通过调控掺杂N的方式和条件,可以有效控制掺杂N基团的生成种类和含量,从而精准实现高活性和稳定性非金属催化剂材料的可控制备。
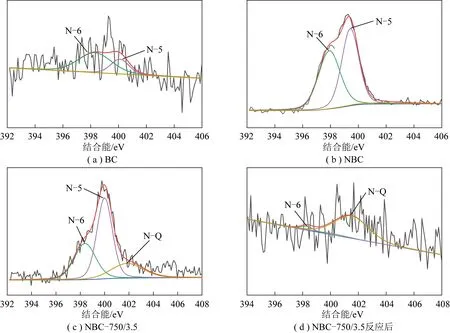
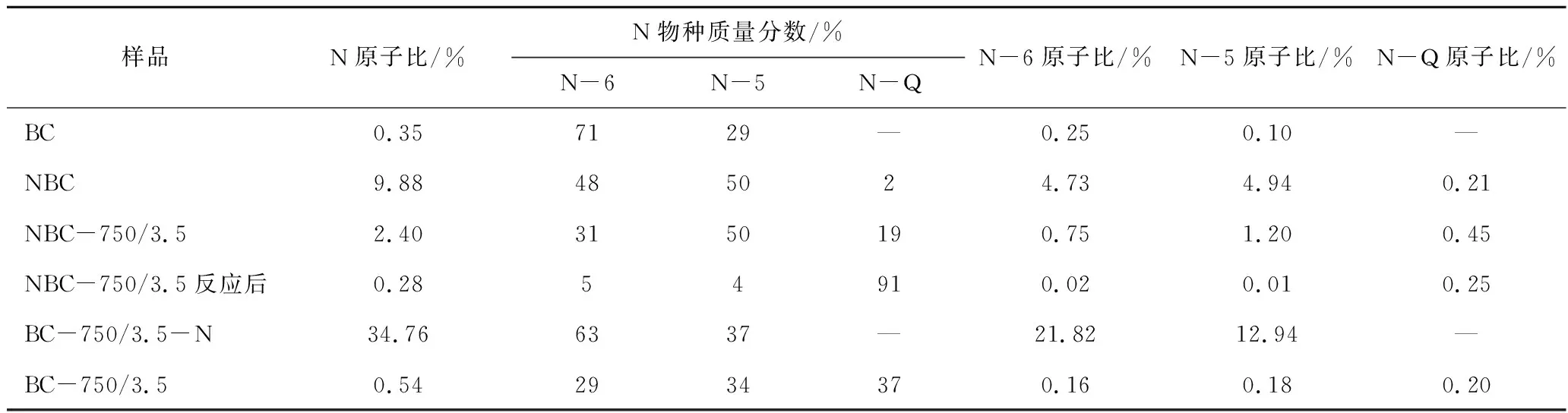
表3 新鲜和反应后催化剂的XPS N1s分析
2.6 CO2-TPD和TG表征分析
图5为不同样品的CO2-TPD图谱。由文献[30-31]可知,在CO2-TPD图谱中,样品特征峰出现的位置温度越高,材料的碱性位越强;其中,位于0~300 ℃的为弱碱性位,位于300~600 ℃的为中等强度的碱性位,温度高于600 ℃的为强碱性位。由图5(a)可知,BC、NBC和NBC-750/3.5三种样品在300~700 ℃均出现1个明显的特征峰,这说明3种样品均含有中、高强度的碱性位,因此其初始活性都较高。根据特征峰宽度和强度的不同,3种不同样品中碱性位排序为:NBC的碱性位最多,NBC-750/3.5次之,BC最少,这与表3中3种样品的N含量测定结果一致。同时也说明,N掺杂有利于增强所制备催化剂材料的碱性位,从而有助于提高催化剂活性和稳定性。BC活化后BC-750/3.5样品的特征峰发生了偏移,表明经过高温活化后一部分碱性位向(左)弱碱性位偏移,一部分向(右)强碱性位偏移。虽然向右偏移增强了碱性位,但由于BC本身的N含量很低(表3),因此其初始活性相对较低。由图5还可知,BC-750/3.5-N样品出现二个较强的特征峰,分别位于250~450 ℃和450~750 ℃ 处。进一步表明,低温掺杂的N保留了较多的碱性位,从而具有较好的稳定性。但由表1可知,由于N掺杂的温度较低,部分N源不能充分分解,从而堵塞了样品的部分孔道,导致比表面积和孔体积均较低,不利于反应气体的质量传输。由图5(b)可知,样品NBC-750/3.5进行10 h催化反应后,其碱性位的强度不仅降低,部分中等强度的碱性位向弱碱性位(100~300 ℃)偏移,催化活性降低;同时部分向强碱性位(600~700 ℃)偏移,其催化稳定性增强。这表明在重整反应过程中,一部分碱性位被消耗,一部分碱性位发生转化。结合前述XPS分析结果可知,样品中的N-5和N-6部分被消耗,部分在反应过程中向N-Q发生转变,其对催化剂的活性和稳定具有明显的相关性。
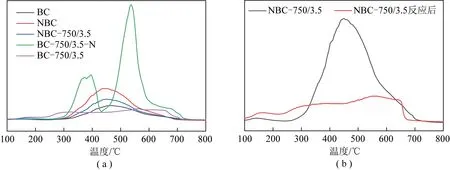
图5 不同新鲜和反应后样品的CO2-TPD图谱Fig.5 CO2-TPD spectrum of fresh and spent catalysts
图6为不同新鲜及反应后样品的TG曲线。由图6(a)可知,所有样品的失重曲线可以分为3个阶段。第1阶段(0~100 ℃),对应于吸附的空气及外在水的脱附[32];第2阶段(100~350 ℃)失重较小,对应于材料内在水以及易挥发物质的挥发;第3阶段(300~600 ℃)失重最多,对应于样品中碳及其活性物质与O发生氧化反应;600 ℃之后样品质量不再变化,即生物质碳在600 ℃之前被氧化完全。这表明重整反应过程中没有生成积碳或生成的积碳也具有较高的反应活性,所制备的碳材料催化剂具有较强的抗积碳性。通过对比还发现,不论是原位掺杂还是后处理掺杂,掺杂N之后的样品NBC-750/3.5和BC-750/3.5-N在第3阶段的失重温度都比未掺杂N的BC-750/3.5样品的失重温度高,而且完全失重最终的温度也明显增加。进一步结合表2和表3可知,样品所含N含量越高,样品失重发生的温度越向高温方向移动。这说明掺杂的N与C的结合提高了所制备材料的稳定性,因此在重整反应过程中也能保持较好的稳定性。由图6(b)可知,3种样品在0~500 ℃基本没有发生失重,其失重区间主要集中在500~650 ℃,650 ℃之后样品被氧化完全,也不再失重。与新鲜样品相比,反应后的样品剧烈失重向高温移动,一方面是由于新鲜材料中易氧化的碳以及具有较强活性的含O、含N官能团在高温重整反应中被消耗;另一方面高温重整反应使部分碳向石墨化转变,同时伴随重整副反应生成高度石墨化的碳,在二者的共同作用下,反应性的催化剂样品表现出更高的抗氧化性。另外,通过对比3种反应后的失重温度可知,反应后NBC-750/3.5样品的完全失重温度最低,BC-750/3.5-N样品次之,BC-750/3.5样品最高,这表明掺杂的N有助于提高碳材料的抗积碳性;同时原位N掺杂由于经过高温活化,与碳形成的结构更为稳定[7]。因此,其不仅具有较好的抗积碳性,而且具有较好的催化活性。
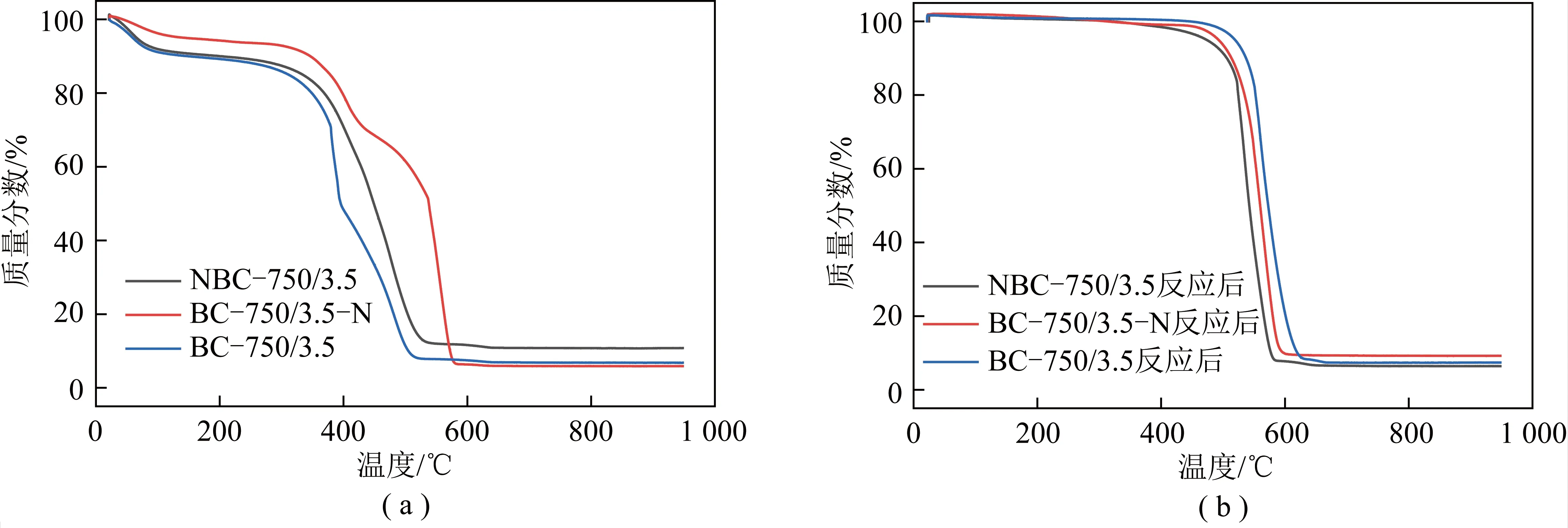
图6 样品的TG曲线Fig.6 TG curves of fresh and spent catalysts
2.7 不同催化剂性能评价
图7和图8为不同催化剂催化CH4-CO2重整性能及稳定性评价曲线(反应条件:900 ℃,CH4和CO2总流量120 mL/min(CH4/CO2=1),2.5 g催化剂)。
由图7可知,所有催化剂的CH4和CO2转化率都随反应时间的延长表现出先下降然后趋于稳定的趋势。由表2可知,生物质含有丰富的含O官能团,具有一定活性[17],能够有效促进CH4-CO2重整反应[18]。BC样品经过7 h反应后,CH4和CO2的转化率稳定在24%和43%左右。但NBC样品的活性较差,初始CH4和CO2转化率仅为11%和28%;而且其稳定性也很差,重整1 h反应后,CH4和CO2的转化率降低到2%和10%。结合前述分析可知,这主要是由于:① 350 ℃掺杂的N源不能完全分解,有效N掺杂效率较低;② 未分解完全的N源物质堵塞了材料孔道,使材料原有的活性位点被覆盖,从而使样品表现出较低的催化活性和较差的稳定性。结合表1可知,BC-750/3.5样品具有较高的比表面积和较大的孔体积,这有利于气体的传质,从而提高样品的催化性能;经过10 h重整反应后,BC-750/3.5样品的CH4和CO2转化率稳定在31%和53%左右。进一步通过后处理掺杂N后,其N含量明显提升,但催化活性反而降低。结合前述分析可知,这主要是由于后处理掺杂材料较小的比表面积和孔体积使得CH4和CO2分子只能在N掺杂生物质碳表面进行吸附,不利于原料气与活性位点的接触[26-27],从而使得BC-750/3.5-N催化剂表现出较低的催化剂活性。但由于掺杂的N含量较高,具有较好的稳定性。由上述分析可知,N掺杂生物质碳重整反应的催化性能由材料的孔结构特性和含N物种的类型和数量共同决定:其大的比表面积和孔体积有利于原料气的吸附和传质;不同含N物种为原料气提供活性位点,有利于气体分子的活化转化。结合前述的表征分析结果可知,原位掺N制备的NBC-750/3.5不仅具有大的比表面积和优异的孔道结构,而且具有较强的碱性位,从而有利于CH4和CO2分子的吸附和活化,表现出最佳的催化活性和稳定性(抗积碳性)。重整反应10 h后,NBC-750/3.5催化剂的CH4和CO2转化率分别稳定在45%和62%,而BC-750/3.5-N的CH4和CO2转化率仅为20%和45%。通过元素分析结果可知,与反应前相比,反应后生物质碳中的N和O含量均下降,说明反应过程中含N和含O官能团被消耗,导致生物质碳材料的催化活性下降。为了进一步考察NBC-750/3.5的潜在应用前景,对其进行了50 h长期稳定性试验,结果如图8所示。由图8可知,进行50 h重整反应后,原位掺杂N制备的催化剂仍能保持较好的稳定性,CH4和CO2的转化率稳定在45%和62%左右;反应后气体的H2/CO基本稳定在0.7左右。
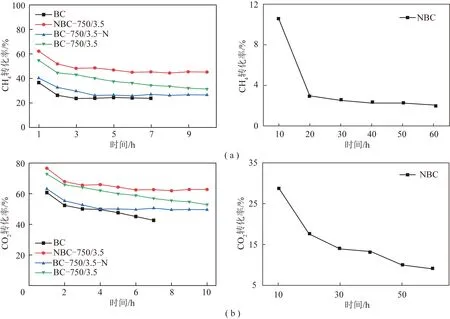
图7 不同催化剂CH4-CO2重整反应性能测试Fig.7 CH4-CO2 reforming reaction performance test of different catalysts
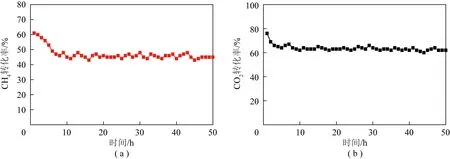
图8 NBC-750/3.5催化剂50 h CH4-CO2重整反应稳定性测试Fig.8 Stability test of NBC-750/3.5 catalyst for 50 h CH4-CO2 reforming reaction
3 结 论
1)生物质先活化,可以形成大量的微孔或窄微孔结构,为后处理掺N提供了良好的孔道结构,从而提高有效N掺杂的含量,制备样品N质量分数最高可达26.35%;但后掺杂制备的样品比表面积和孔体积较低,不利于反应气体的传递和吸附,因此表现出较差的催化性能。
2)在进行原位N掺杂活化时,活化剂和掺杂的N产生相互协同作用,更有利于造孔作用,从而提高了制备NBC-750/3.5样品的比表面积和孔体积,有利于反应气体的传递和吸附。
3)生物质原位N掺杂不仅提高了其比表面积和孔体积,而且引入了丰富的含N碱性官能团,其主要以吡啶N(31.15%)、吡咯N(49.98%)和石墨N(18.86%)3种形式存在;较高的吡啶N和吡咯N占比,有利于催化剂的活性和稳定性(抗积碳性);经过50 h重整反应后,CH4和CO2的转化率稳定在45%和62%左右。
4)进行N掺杂形成的吡咯N有利于CO2吸附,吡啶N有利于分子活化。因此,通过调控催化剂的制备方法和N杂原子掺杂方式,不仅可以更准确地调控含N基团的生成类型,而且通过调控可获得高性能非金属碳基非再生型重整催化材料。
猜你喜欢
家庭科学·新健康(2022年7期)2022-07-13
银行家(2022年5期)2022-05-24
表面技术(2022年1期)2022-02-12
福建农林大学学报(哲学社会科学版)(2021年5期)2021-12-04
法制博览(2021年14期)2021-11-25
市场周刊(2021年8期)2021-11-22
装备维修技术(2020年5期)2020-11-20
矿产综合利用(2020年1期)2020-07-24
伴侣(2018年8期)2018-08-23
中小企业管理与科技·上旬刊(2018年12期)2018-02-18
