
聚乙烯己内酰胺链端改性及其对甲烷水合物形成的抑制作用研究
2022-05-26 02:57:38唐翠萍张雅楠梁德青李祥
化工学报 2022年5期
唐翠萍,张雅楠,4,梁德青,李祥,5
(1中国科学院广州能源研究所,广东广州 510640; 2中国科学院天然气水合物重点实验室,广东广州 510640;3广东省新能源和可再生能源研究开发与应用重点实验室,广东广州 510640; 4中国科学院大学,北京 100049;5中国科学技术大学,安徽合肥 230026)
引 言
天然气水合物是水与小分子气体在高压低温条件下形成的似冰状的非化学计量的笼形晶体物质,主体水分子通过氢键相连形成多面体笼孔,客体分子(如甲烷、乙烷、二氧化碳和硫化氢等)填充在这些笼孔中形成不同类型的气体水合物[1-2]。由于晶体结构的不同,可将水合物分为三类:Ⅰ型、Ⅱ型和H 型[3]。在石油和天然气生产、加工和钻探过程中,由于满足高压低温条件,水与碳氢化合物会形成固态水合物,这不仅会造成管道堵塞、开采设备失灵等安全事故,而且有可能造成巨大的经济损失和环境破坏[4-5]。
油气工业使用多种方法来防止水合物的形成,例如控制压力法、管道加热法以及注入水合物抑制剂,其中加入水合物抑制剂是最常用和有效的水合物预防方法[6]。根据抑制机理的不同主要可分为热力学抑制剂和低剂量水合物抑制剂,传统的热力学抑制剂如甲醇、乙二醇和电解质水溶液等,它们通过改变水溶液或水合物的化学势,将水合物的形成曲线移动到较低温度或较高压力的区域来发挥作用,用量一般占水相的10%~50%(质量),添加量大且经济成本高,最常用的热力学抑制剂甲醇有一定的毒性,会对环境产生不利的影响[7-8]。低剂量水合物抑制剂包括动力学抑制剂和阻聚剂,阻聚剂通常是一些表面活性剂,使用过程中通常需要油相,在天然气管道中动力学抑制剂更为常用[7,9]。
自20 世纪90 年代起,人们开发了多种水合物动力学抑制剂,其通过延迟气体水合物的成核和生长来抑制水合物的生成,用量可低至0.01%~5%[10],动力学抑制剂主要是一些水溶性聚合物,例如聚乙烯基吡咯烷酮(PVP)和聚乙烯基己内酰胺(PVCap)以及它们的共聚物或均聚物,聚乙烯基吡咯烷酮作为第一代水合物抑制剂,其长链上的五元环吸附到水合物表面未完成的孔穴中,阻止了水合物晶核的生长[11],抑制效果在相对较高的温度(12℃)才有效[12],当温度降低到4℃时不再有抑制效果[13]。第二代水合物抑制剂主要有七元环的聚乙烯基己内酰胺(PVCap)、聚(N-乙烯基吡咯烷酮/N-乙烯基已内酰胺)[Poly(VP/VC)]和聚(N-乙烯基吡咯烷酮/N-乙烯基已内酰胺/N,N-二甲胺甲基丙烯酸乙酯)(VC-713)[14]等。聚乙烯基己内酰胺(PVCap)和聚乙烯基吡咯烷酮(PVP)都是内酰胺基类的聚合物,内酰胺环上碳原子数的不同使得PVCap 比PVP 具有更好的天然气水合物抑制性能[15],已有的研究表明,在8~9℃的过冷度下,添加0.5%(质量)的聚乙烯基己内酰胺(PVCap)可以将水合物成核延迟至约24 h,但PVCap 在高于12℃的过冷度下会失去抑制效果[12]。虽然PVP 和PVCap 等能有效抑制水合物的形成,但仍难以满足所有条件下的工业要求,应用还不够广泛,因此有必要对聚乙烯基吡咯烷酮(PVP)和聚乙烯基己内酰胺(PVCap)进行改性使其具有更好的抑制性能。
利用计算机分子模拟技术发现,在抑制剂中引入亲水或者疏水性基团能有效改善抑制剂的抑制性能[16-17],如Zhang 等[18]将亲水性的羧基引入聚乙烯基己内酰胺(PVCap)分子链末端对其进行改性合成了新型水合物抑制剂PVCSCOOH,其对甲烷水合物的抑制性能强于PVCap。Kelland等[19]将疏水性的烷基如甲基、异丙基等引入聚乙烯基吡咯烷酮的内酰胺环上合成一系列1-烷基-乙烯基吡咯烷酮(1-烷基-VPs)的水溶性均聚物和共聚物。此外,将N-乙烯基吡咯烷酮(VP)或N-乙烯基己内酰胺(VCap)单体与其他单体进行共聚也能有效改善抑制剂的抑制性能,如Lou 等[20]将(乙二醇)甲基醚甲基丙烯酸酯单体与N-乙烯基己内酰胺单体共聚合成聚(环氧乙烷-co-乙烯基己内酰胺)(PEO-co-VCap-1)。Long 等[21]将N-乙烯基己内酰胺单体与1-乙烯基咪唑共聚得到共聚物PVCap-co-VIM,新型抑制剂PVCap-co-VIM 比PVPK90 更有效延迟水合物晶体成核。Liang 等[22]在聚乙烯己内酰胺分子链端引入—S(CH2)2OH基团,提高了聚合物对甲烷水合物的抑制性能。
目前很少有关于在聚乙烯基己内酰胺(PVCap)分子链中引入两种基团对抑制甲烷水合物生成方面性能的探究,本文以聚乙烯基己内酰胺(PVCap)为基础,将氧乙基和酯基分别引入聚乙烯基己内酰胺(PVCap)分子链端合成改性抑制剂PVCap-XA1,并采用FTIR和1H NMR方法对合成的改性抑制剂进行表征,使用恒容恒温法和匀速降温法评价改性抑制剂PVCap-XA1对甲烷水合物的抑制性能,通过粉末X 射线衍射(PXRD)、低温激光拉曼光谱和冷冻扫描电子显微镜(Cryo-SEM)研究改性抑制剂对水合物晶体结构和形貌的影响。
1 实验材料和方法
1.1 材料
乙基黄原酸钾,纯度98%,上海麦克林公司;2-溴丙酸甲酯,纯度87%,上海麦克林公司;乙醇,纯度大于99.7%,上海阿拉丁生化科技股份有限公司;二氯甲烷,纯度大于99.8%,上海麦克林公司;灭菌水,纯度大于99.99%广州鼎国生物技术有限公司;无水硫酸镁,纯度99.99%,上海麦克林公司;N-乙烯基己内酰胺(NVCap),纯度大于98.0%,上海梯希爱化成工业公司;1,4-二氧六环,纯度大于99.5%,上海阿拉丁生化科技股份有限公司;2,2-偶氮二异丁腈(AIBN),纯度大于98.0%,上海梯希爱化成工业公司;正己烷,纯度97%,上海麦克林公司;甲烷气体,纯度大于99.0%,广州市粤佳气体有限公司;水为实验室自制的蒸馏水。
1.2 抑制剂合成
链转移剂XA1的合成:将0.020 mol乙基黄原酸钾溶解在25 ml 乙醇中,完全溶解后,加入2 ml 2-溴丙酸甲酯,并将溶液室温搅拌反应18 h,过滤除去白色沉淀物,剩余溶液置于旋转蒸发仪中85℃抽真空旋蒸大部分溶剂,冷却后向产物中加入25 ml 去离子水,并将浓缩物转移至分液漏斗中,加入50 ml 二氯甲烷并剧烈摇动,回收有机层丢弃水层,重复三次后用无水MgSO4去除过量的水,过滤去除无水MgSO4,蒸发二氯甲烷后获得黄色油状液体[合成路线见图1(a)]。产率为65.7%。
改性抑制剂PVCap-XA1和PVCap的合成:在圆底烧瓶中将1,4-二氧六环(5 ml)、NVCap(13.92 g,0.1 mol)和AIBN(16.67 mg,0.1 mmol)与XA1(50 mg,0.24 mmol)混合,密封后抽真空、通氮气反复3 次以保证隔绝空气,在氮气保护下置于油浴中于60℃加热反应约6 h,通过将烧瓶置于2℃冰箱中20 min 来淬灭反应,然后通过在己烷中沉淀来纯化产物得到PVCap-XA1[合成路线见图1(b)],合成物产率69.7%。PVCap 是在相同实验条件下合成的,但不添加链转移剂XA1[合成路线见图1(c)],产率为63.2%。
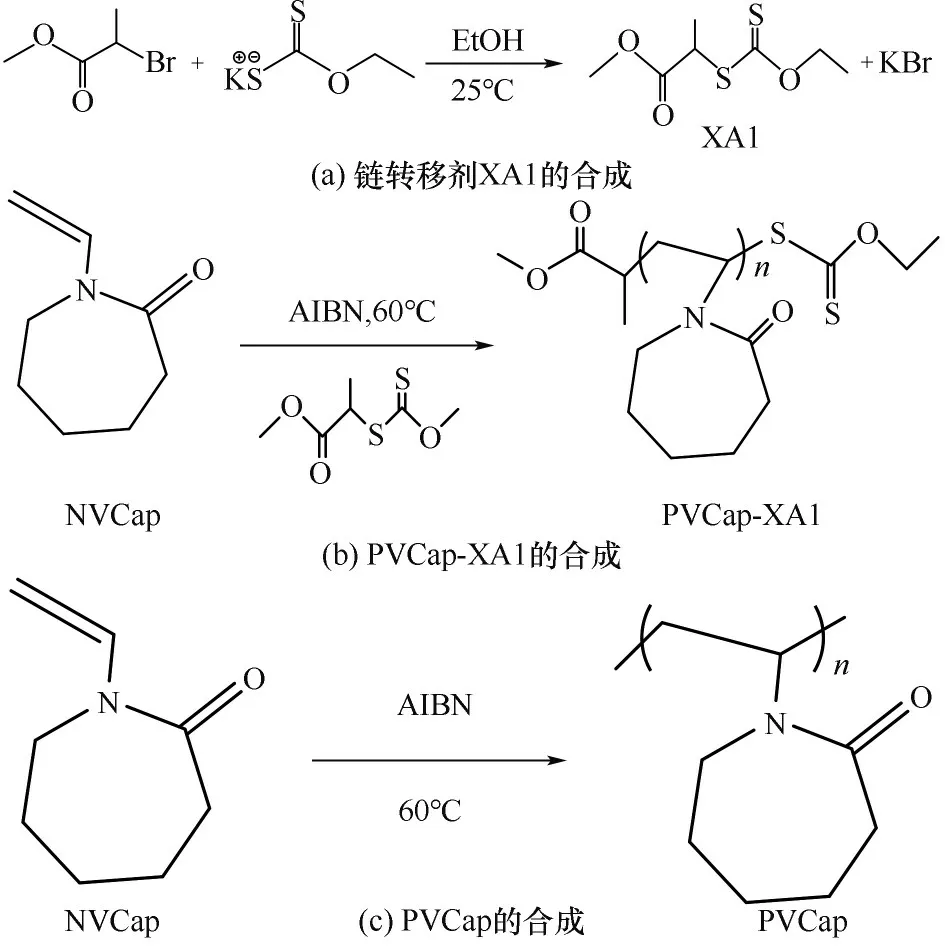
图1 XA1、PVCap和PVCap-XA1的合成Fig.1 The synthesis of XA1,PVCap and PVCap-XA1
1.3 抑制性能测试
1.3.1 实验装置 恒温恒容法测甲烷水合物生成诱导时间的实验装置[22],主要包括一个高压不锈钢反应釜,恒温水浴,预冷气体缓冲罐,磁力搅拌器,数据采集系统(每10 s采集一次)和用于记录数据的计算机。反应釜有效容积为100 ml,承受最大工作压力为20 MPa。
恒容匀速降温法测甲烷水合物形成最大过冷度的实验装置如图2 所示,实验装置主要包括容量为40 ml 和额定工作压力为12 MPa 的蓝宝石反应釜,磁力搅拌转速为0~1200 r/min;压力传感器型号 为CYB-20S,测 试 范 围 为0~20 MPa,精 度为±0.025 MPa;温度传感器型号为PT100,测试范围为-40~100℃,精度为±0.1℃;反应釜置于WGD/J-108高低温试验箱中。

图2 水合物动力学抑制实验装置示意图Fig.2 Schematic illustration of the kinetic hydrate inhibition experimental apparatus
1.3.2 实验过程 恒温恒容法可以测量抑制剂溶液的诱导时间。首先,将装有30 ml 抑制剂溶液的不锈钢高压反应釜放入4℃的恒温浴中,待温度稳定在4℃后,抽真空通入8 MPa 的甲烷气体,关闭气阀,待温度和压力稳定后立即启动磁力搅拌器(转速为1000 r/min),诱导时间(ti)定义为搅拌起始点与压力立即开始下降(第一个水合物晶体成核)之间的时间间隔。
恒容匀速降温法用于测量抑制剂溶液的最大过冷度。将10 ml 抑制剂溶液放入蓝宝石反应釜中,抽真空后通入9 MPa 的甲烷气体,关闭气阀,打开磁力搅拌器(转速为900 r/min),将空气浴装置以1℃/h 的恒定冷却速度从+20℃冷却至-10℃以形成水合物,在降温过程中出现压力骤降和温度骤升的现象表明甲烷水合物生成,将水合物形成的起始温度记为To,最大过冷度是水合物平衡温度和To之间的差值。
1.3.3 水合物样品的制备 将30 ml 抑制剂溶液放入不锈钢高压反应釜中,置于4℃恒温水浴中,待温度稳定,抽真空后通入10 MPa 的甲烷气体,关闭气源,启动磁力搅拌器(转速为1000 r/min),观察到压力骤降、温度骤升说明水合物大量生成,之后反应釜内压力保持稳定,继续放置一周左右。一周后,将高压反应釜从恒温水浴箱中取出并快速擦干表面水分,随后放入装有液氮的保温桶中进行快速冷冻,2 min 后打开高压反应釜,并向其中不停地加入液氮以防止水合物分解。盛放水合物的冻存管提前在液氮中冷冻,然后在液氮保护下取出反应釜中的水合物样品装于冻存管中,再保存在液氮罐中。进行粉末X 射线衍射测试时,将研钵提前放入液氮中冷冻,一段时间后,将水合物样品从冻存管中取出倒入装有液氮的研钵中研磨成粉末,在液氮保护下,快速转移至X 射线衍射仪样品台中的凹槽中进行水合物晶体结构的测试;进行拉曼测试时,取出冻存管放在装有液氮的研钵中,将水合物颗粒研磨成1 mm直径大小的颗粒,在氮气保护下将水合物颗粒转移至拉曼光谱仪的石英坩埚中,旋紧盖子,在液氮吹扫下进行测试;低温扫描电镜测试时,取出水合物放入装有液氮的研钵中研磨至“米粒”大小,在氮气保护下,将水合物颗粒放入提前折叠好的锡纸凹槽中,包裹紧实后,放入冷冻输送系统进行前处理,拨开水合物样品后进行拍摄。
1.4 结构表征
采用布鲁克公司TENSOR27型傅里叶变换红外光谱仪对PVCap-XA1、PVCap 和单体NVCap 进行FTIR 测试,溴化钾压片;采用布鲁克公司AVANCEⅢ型核磁共振仪对PVCap-XA1、PVCap 进行1H NMR 测试,氘代氯仿溶剂。甲烷水合物的晶体结构由粉末X 射线衍射仪(X’Pert Pro MPD,PANalytical)在-50℃和常压203 K条件下测定,2θ范围为5°~80°,水合物笼占据特性通过激光共聚焦拉曼光谱仪(Lab RAM HR800,Horiba)在-50℃和常压203 K 条件下测量,其激发源532 nm Ar+,具有50 mW 的强度,气体水合物的晶体形态通过低温扫描电镜(Hitachi,S-4800)在-100℃真空环境下获得。
2 实验结果与讨论
2.1 XA1、PVCap-XA1和PVCap的结构表征
2.1.1 FTIR 表征结果 图3 为NVCap、PVCap 和PVCap-XA1 的FTIR 谱 图,由 图3 可 知,波 数 为1080 cm-1和1195 cm-1的吸收峰归属于己内酰胺环上C—N 的弯曲振动;波数为1626 cm-1的吸收峰属于内酰胺环上C==O 键的伸缩振动[23-24];波数为1665 cm-1的吸收峰属于单体NVCap 中C==C 键的伸缩振动峰;波数为3109 cm-1的吸收峰属于C—H 键的伸缩振动峰[22],NVCap、PVCap 和PVCap-XA1 均出现内酰胺环上C==O 键的伸缩振动吸收峰、C—N单键的弯曲振动吸收峰,说明3 种物质都具有PVCap 的 基 本 结 构,且PVCap 和PVCap-XA1 在1665 cm-1(υC==C)和3109 cm-1(υC—H)处的吸收峰消失,说明PVCap 和PVCap-XA1 聚合成功,PVCap-XA1 的红外谱图中,波数为1046 cm-1处的吸收峰属于“CS2”整体的伸缩振动峰。聚合物中3447 cm-1处的吸收峰为缔合羟基(O—H)的伸缩振动吸收峰,也可能由聚合物的亲水性所致。
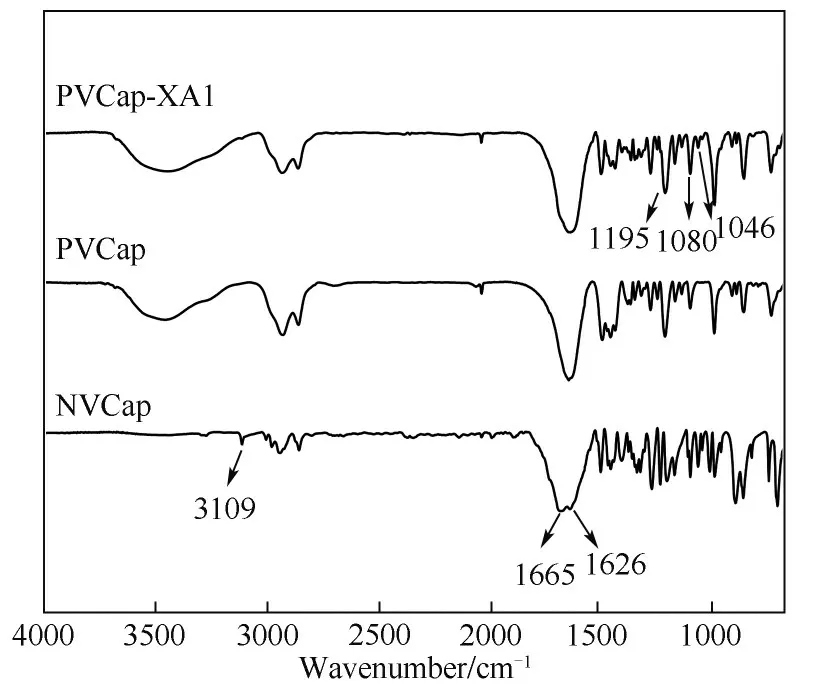
图3 NVCap、PVCap和PVCap-XA1的FTIR谱图Fig.3 FTIR spectra of NVCap,PVCap and PVCap-XA1
2.1.21H NMR 表 征 结 果 图4 为XA1、PVCap 和PVCap-XA1 的1H NMR 图。由 图4 XA1 的1H NMR图可知:δ=1.41(3H,t,—OC2H5的—CH3),1.57(3H,d,—CHCH3S—的—CH3),3.75(3H,s,—OCH3的—CH3),4.38(H,q,—CHCH3S—的—CH)和4.62(2H,q,—OCH2CH3的—CH2)。由图4 PVCap-XA1的1H NMR 图 可 知,δ=4.25~4.65 为—NCH—、—OCH2—和—CHCH3—的—CH—的质子峰(“7、8、10”),δ=3.65 为—OCH3的质子峰(“12”),δ=2.88~3.46 为—NCH2—的质子峰(“1”),δ=2.23~2.7 为—COCH2—的质子峰(“5”),δ=1.5~2.0 为己内酰胺环上—CH2—的质子峰(“2、3、4”),δ=1.0~1.5为主链上—OCH2CH3中—CH3、—CHCH3—的—CH3和主链上—CH2—的质子峰(“6、9、11”)。由图4 PVCap的1H NMR 图可知,δ=2.88~3.46 为—NCH2—的质子峰(“a”),δ=1.0~2.0 为己内酰胺环上—CH2—和主链上—CH2—的质子峰(“b、c、d、g”),δ=2.23~2.7为—COCH2—的 质 子 峰(“e”),δ=4.25~4.65 为—NCH—的质子峰(“f”)。对比PVCap 和PVCap-XA1 的1H NMR 图可知两者都具有PVCap 的基本结构,且PVCap-XA1 的1H NMR 图中δ=3.65 为甲基丙烯酸甲酯中—COOCH3官能团中的质子峰,再结合PVCap-XA1 的红外谱图中波数为1046 cm-1处的吸收峰属于“CS2”整体的伸缩振动峰,证明PVCap-XA1成功合成。
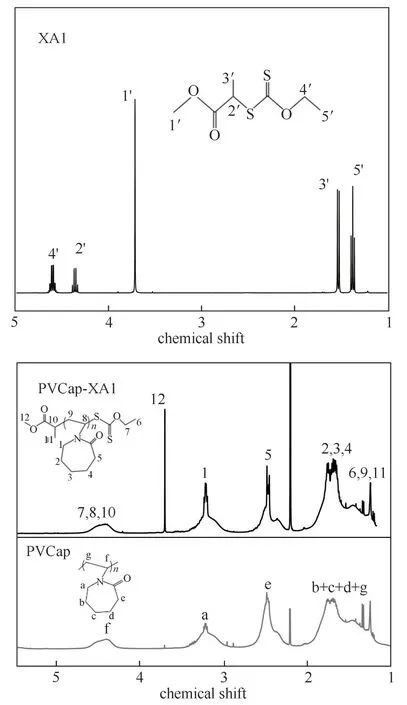
图4 XA1、PVCap-XA1 和PVCap的1H NMR图Fig.4 1H NMR spectra of XA1,PVCap-XA1 and PVCap
2.2 PVCap-XA1对甲烷水合物形成的抑制作用
2.2.1 诱导时间 恒温恒容法用于测量抑制剂溶液的诱导时间,图5 为添加2.0%(质量)PVCap-XA1抑制剂和纯水情况下水合物生成的温度、压力随时间的变化曲线。由图5 可知,含2.0%(质量)PVCap-XA1 抑制剂的水合物生成体系,刚打开搅拌开关时压力下降约0.1 MPa、温度略微上升,这是由于部分甲烷气体溶于水中。此后温度和压力保持恒定,240 min 后出现压力突降、温度骤升,表明水合物大量生成,大量甲烷气体与水结合生成甲烷水合物,水合物生成为放热反应导致体系温度升高。而在甲烷-纯水体系下,开启搅拌,水合物即刻形成,诱导时间(ti)几乎为0,这表明2.0%(质量)PVCap-XA1对甲烷水合物的生成具有明显的抑制作用。
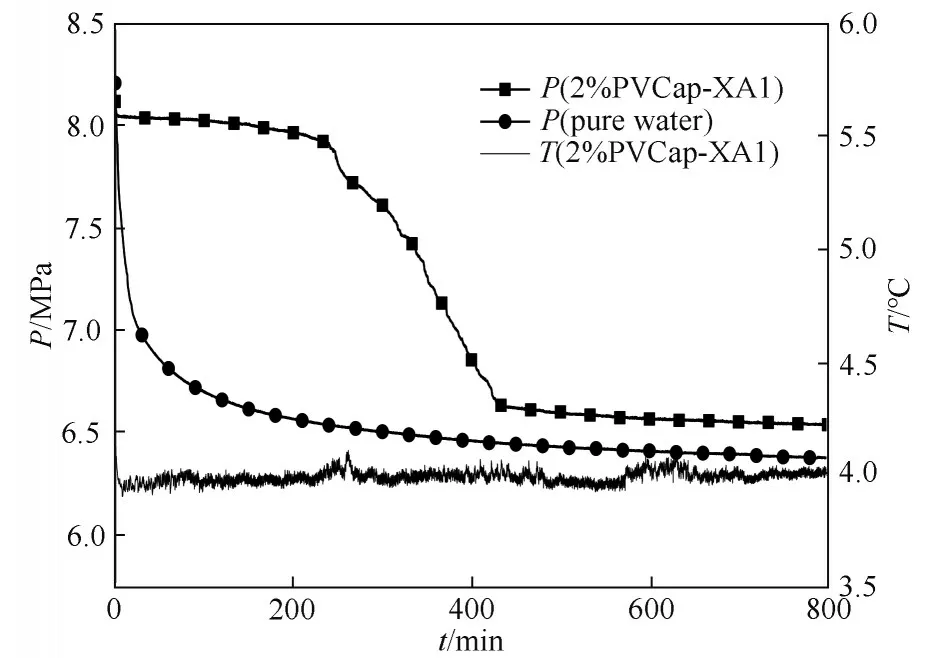
图5 含2.0%(质量)PVCap-XA1抑制剂溶液以及纯水体系水合物生成温度、压力曲线Fig.5 Pressure-temperature curves of hydrate formation with 2.0%(mass)PVCap-XA1 and without inhibitors
2.2.2 最大过冷度 恒容匀速降温法用于测试含抑制剂溶液的最大过冷度,过冷度反映了在水合物生成之前,系统可以冷却到低于水合物形成的平衡温度的程度,最大过冷度定义为水合物平衡温度和To之间的差值[25],抑制剂抑制能力越强,水合物形成温度偏离相平衡温度越大,最大过冷度也就越大。图6为恒定冷却法测得含2.0%(质量)PVCap-XA1抑制剂的水合物生成的典型曲线。刚开始搅拌时,反应釜内的压力和温度均以恒定的速率下降,约1460 min后,由于水合物形成,导致反应釜内的压力突降,但是由于水合物生成量不多以及水合物形成的热量被迅速导走,导致温度只是略微升高。水合物起始形成温度为发生急剧压降拐点处的温度,记为To。最大过冷度即为此时压力(Po)对应的水合物平衡温度与To之间的差值。含2.0%(质量)PVCap-XA1 抑制剂的水合物形成温度To为0.9℃,对应的最大过冷度为10.5℃。
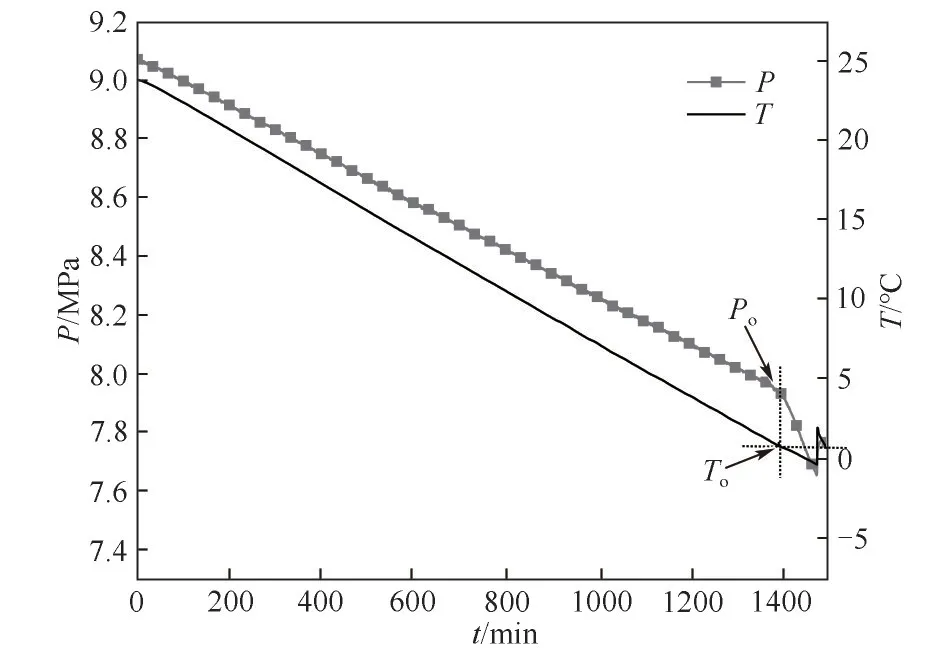
图6 恒定冷却法测含2.0%(质量)PVCap-XA1抑制剂的水合物生成的示例曲线Fig.6 Typical curve of hydrate formation with 2.0%(mass)PVCap-XA1 through constant cooling test
表1 列 举 了PVCap 和PVCap-XA1 在0.5%、1.0%和2.0%(质量)浓度下的水合物生成温度To和最大过冷度,其中纯水样品(未添加动力学抑制剂)作为空白对照组,由表可知,纯水中水合物生成温度为8.8℃,过冷度为3.0℃,这与之前文献报道的数据一致[18,26]。而添加了不同质量分数的PVCap 或PVCap-XA1 抑制剂的体系水合物的过冷度与纯水体系相比显著提高,如含2.0%(质量)PVCap-XA1 体系水合物的最大过冷度为10.5℃,与纯甲烷水合物体系相比,最大过冷度提高了7.5℃,说明PVCap 和PVCap-XA1均能有效抑制甲烷水合物的生成。图7为不同浓度下PVCap-XA1 和PVCap 的最大过冷度,由图7可知,最大过冷度受两种聚合物的浓度影响,随着浓度的增加呈上升趋势,但对PVCap-XA1的影响要比PVCap 小,当浓度从1.0%(质量)增加到2.0%(质量)时,PVCap-XA1 的最大过冷度增加了1.4℃,而PVCap 的最大过冷度增加了2.2℃。对于相同浓度 的PVCap 和PVCap-XA1 体 系,PVCap-XA1 比PVCap 提供了更高的最大过冷度,特别是在浓度为1.0%(质量)时,PVCap-XA1 比PVCap 高3.4℃,即相同浓度下PVCap-XA1比PVCap抑制能力好。
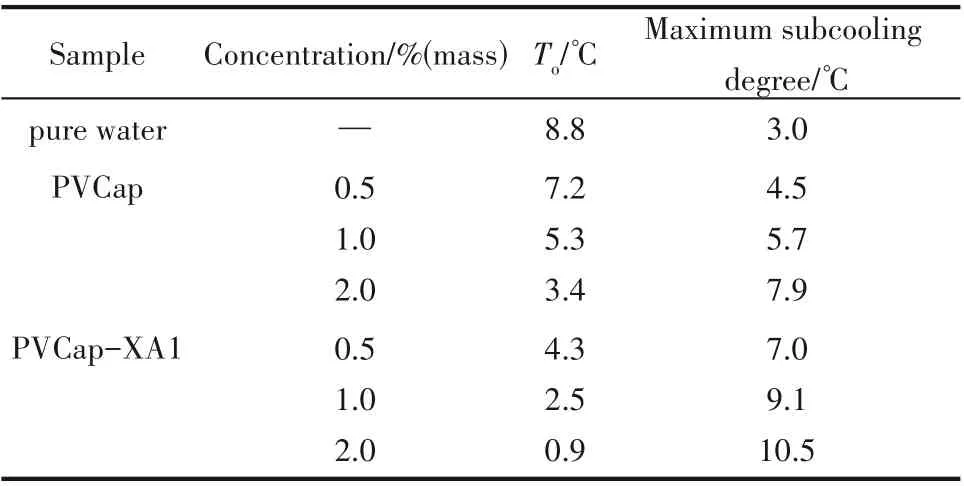
表1 PVCap和PVCap-XA1在不同浓度下的To和最大过冷度Table 1 Toand maximum subcooling of PVCap and PVCap-XA1 at different concentrations
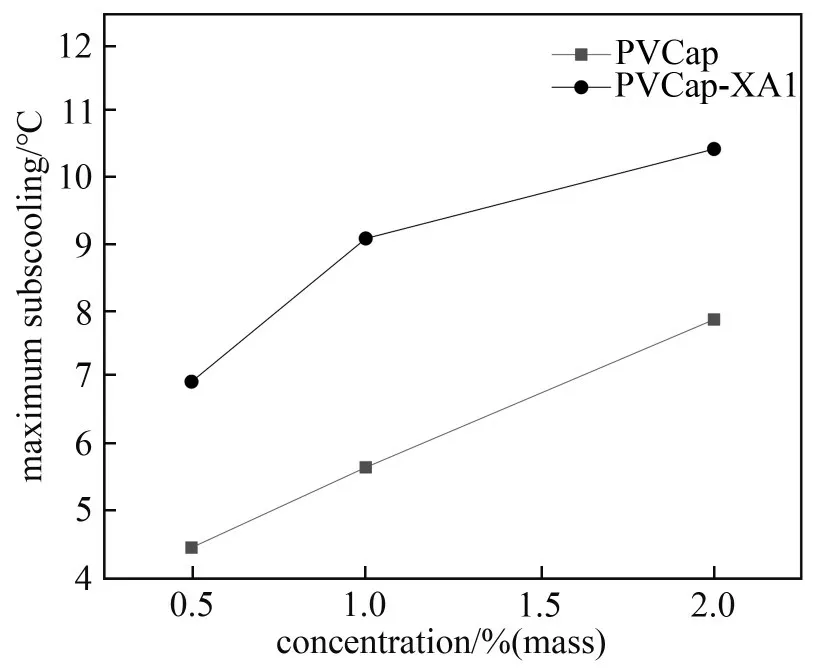
图7 不同浓度PVCap-XA1和PVCap 的最大过冷度Fig.7 Maximum subcooling degree of PVCap-XA1 and PVCap versus concentration
表1中所采用的数据为三组重复数据中最接近平均值的实验组数据。以改性抑制剂为例,进行数据误差分析,图8给出了重复实验的最大过冷度数据和平均值偏差。从图8可以看出表1采用的测量数据和平均值很接近。以绝对误差来说,误差最大出现在浓度0.5%(质量)时,和平均值相差0.9℃,此时相对误差为12.9%,其他数据的相对误差均在3.5%以内,测量值和平均值均差距不大,可以认为数据可靠。
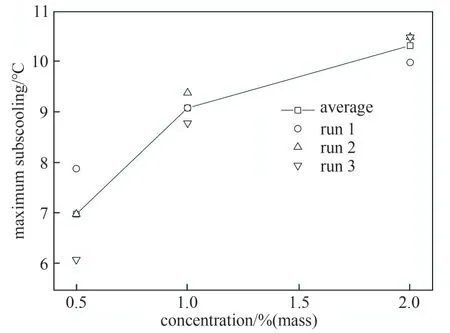
图8 不同浓度PVCap-XA1最大过冷度偏差Fig.8 Maximum subcooling deviation of PVCap-XA1 at different concentrations
2.3 PVCap-XA1对甲烷水合物结构的影响
多晶X 射线衍射可测定甲烷水合物的晶体结构,图9 为添加2.0%(质量)PVCap-XA1 抑制剂和不添加抑制剂(纯水)情况下体系生成的甲烷水合物的PXRD 图,甲烷水合物为Ⅰ型水合物[27],在2θ为27.3°、28.4°、30.4°、31.4°、32.3°处的衍射峰分别代表sI 水合物的(3 2 0)、(3 2 1)、(4 0 0)、(4 1 0)和(4 1 1)晶面[28]。图9 中标记“*”的衍射峰为其冰峰,标记数字的衍射峰为甲烷水合物的特征峰[29],对比纯水和2.0%(质量)PVCap-XA1 体系生成的水合物的PXRD图,发现添加PVCap-XA1抑制剂的水合物峰位置与纯甲烷水合物峰位置完全重叠,说明PVCap-XA1的加入在动力学实验上抑制了甲烷水合物的生成但并未改变其晶体结构。X 射线衍射的峰强度与晶胞内原子类型、原子数目和原子的位置有关,纯水峰强比较大,说明测试时用的纯水样品中的水合物纯度较高。为了不让样品的特殊性影响实验结果,实验选用同次样品内的相对峰强进行分析。但在纯水-甲烷体系中,甲烷水合物在(3 2 1)和(3 2 0)晶面的晶峰强度比值为2.6,大于其在2.0%(质量)PVCap-XA1 抑 制 剂 体 系 中 的 值(2.15),说 明PVCap-XA1抑制剂选择性地作用在特定的晶面上,改变了(3 2 1)和(3 2 0)晶面的相对峰强度。

图9 2.0%(质量)PVCap-XA1和纯水体系存在下形成的甲烷水合物的PXRD图Fig.9 PXRD patterns of CH4 hydrate formed in the presence of 2.0%(mass)PVCap-XA1 or pure water
拉曼光谱可测得客体分子在气体水合物中的分布,图10 为纯水和不同浓度PVCap-XA1 体系存在下形成的甲烷水合物的拉曼谱图,纯甲烷水合物的拉曼谱图在波数为2905 cm-1处有一个较高的峰,在波数为2915 cm-1处有一个较低的峰,它们分别对应着甲烷气体在水合物笼中的大笼(51262)和小笼(512)的v1对称振动带[30-31],比较添加不同浓度的PVCap-XA1 体系和纯水体系的峰位置发现没有明显的差异,表明PVCap-XA1没有改变甲烷水合物的晶体结构,更加支持了PXRD图中得到的结论。

图10 纯水和不同浓度PVCap-XA1体系存在下形成的甲烷水合物的Raman谱图Fig.10 Raman spectra of CH4 hydrates in the presence of PVCap-XA1 with different concentration and pure water
大小笼占比(IL/IS)为大笼(51262)与小笼(512)水合物笼峰强度的化学计量比,其理论值为3.02[32-33],本实验测得纯水-甲烷体系中大小笼占比IL/IS=2.97,接近理论值。计算0.5%、1.0% 和2.0%(质量)PVCap-XA1体系的大小笼占比,发现其值均小于纯水,分别为2.88、2.71 和2.54。计算结果说明,在甲烷水合物形成过程中,游离的甲烷分子优先填充小笼,这与文献报道一致[33],主要是因为小笼中CH4的结合能(Eb=-8.10 kcal/mol,1 cal=4.18 J)明显大于大笼中(Eb=-5.28 kcal/mol),所以甲烷分子优先填充小笼,而添加了PVCap-XA1抑制剂后甲烷在水合物笼中IL/IS比值较低的主要原因是PVCap-XA1 的存在阻碍了甲烷气体的传输,PVCap-XA1通过氢键吸附到水合物表面后,产生的空间位阻效应阻止了甲烷分子通过笼面进入水合物孔穴,甲烷分子倾向于进入更稳定的小笼来稳定水合物,导致更多的大笼变空[33],从而影响了水合物晶笼的稳定性,抑制了水合物的形成。
2.4 PVCap-XA1对甲烷水合物形态的影响
低温扫描电镜可测得水合物的微观形貌,图11为纯水和添加2.0%(质量)PVCap-XA1 体系下生成的水合物晶体形态。由图11可知,纯甲烷水合物晶体形貌呈现出致密且规则的孔,这是亚微米孔隙的典型特征[34-35],在纯甲烷水合物晶体表面出现的多孔状的结构,非常有利于甲烷气体进入水合物笼中从而生成更多的水合物。在添加2.0%(质量)PVCap-XA1 抑制剂后,发现水合物晶体形貌呈现“鱼鳞”状,且水合物晶体颗粒层层叠加,这有可能形成非常致密且“厚重”的结构,从而阻碍甲烷气体进入水合物笼中,起到抑制效果。

图11 纯水和添加2.0%(质量)PVCap-XA1形成甲烷水合物的电镜图Fig.11 Microstructure pictures of the methane hydrate generated in the presence of 2.0%(mass)PVCap-XA1 and pure water
2.5 PVCap-XA1 对甲烷水合物形成的抑制机理分析
对比PVCap-XA1 和PVCap 的化学结构,如图1(b)、(c)所示,PVCap-XA1和PVCap都具有亲水性很强的酰胺基团,在水合物形成之前,亲水性的酰胺基团通过与水合物表面的水分子形成氢键而吸附在水合物晶体表面,随着吸附在水合物表面的聚合物分子越来越多,水合物晶体会发生形变,抑制剂也会在水合物笼中形成空间位阻作用,阻碍了其他甲烷分子进入水合物笼,从而有效延缓了水合物成核和生长[36-37]。PVCap-XA1 比PVCap 抑制水合物生成的性能更好,可归因于PVCap-XA1聚合物主链上含有疏水性的酯基和烷氧基,已有的文献证明动力学抑制剂中含有疏水性的部分有利于提高抑制剂的抑制性能[38],PVCap-XA1 中疏水性的酯基和烷氧基破坏了构成水合物笼型结构的水分子之间的氢键网络,增强了对水结构的扰动,从而延缓了甲烷水合物的成核和生长[39],Cohen 等[40]在研究复合抑制剂(KHIs和AAs联用)的抑制机理时认为,疏水性的烷氧基使抑制剂分子链的构象得以扩展,增加其可用表面积,使水合物的笼表面与抑制剂分子能更充分地相互作用。当PVCap-XA1分子溶解时,其中酰胺基团通过氢键吸附在水合物表面,PVCap-XA1 另一端主链上疏水性的酯基和烷氧基可以阻止游离的水分子靠近水合物笼,阻碍了水合物笼的形成,进而延缓了水合物生成速度。此外,PVCap-XA1 的高分子结构还可以与游离的水分子发生氢键相互作用,使用于形成水合物笼的水分子减少,进而阻碍水合物成核和生长。
3 结 论
本文以现有抑制剂PVCap 结构为基础,通过氧乙基和酯基在其端链进行改性,合成了PVCap-XA1,并研究了PVCap-XA1 对甲烷水合物的抑制性能、结构和微观形貌的影响。
(1)实验条件下,与纯水-甲烷系统相比,含2.0%(质量)PVCap-XA1抑制剂的体系能将水合物生成时间延长至240 min;与含相同浓度的PVCap 相比,含PVCap-XA1甲烷水合物形成具有更高的最大过冷度,即PVCap-XA1对甲烷水合物抑制作用更好。这可能是因为PVCap-XA1分子链上存在疏水性的酯基和烷氧基扰乱了构成水合物笼型结构的水分子之间的氢键网络,从而延缓了水合物的成核和生长。
(2)添加PVCap-XA1并没有改变甲烷水合物的晶体结构,但改变了(3 2 1)和(3 2 0)晶面的相对峰强度,说明PVCap-XA1对水合物笼的晶面作用具有选择性。在PVCap-XA1 存在下,甲烷水合物中大(51262)和小(512)水合物笼甲烷峰强度化学计量比(IL/IS)降低,即抑制剂PVCap-XA1阻止了甲烷分子进入水合物大笼,从而抑制甲烷水合物的形成。
(3)通过低温扫描电镜分析发现,PVCap-XA1加入改变了甲烷水合物的生成形貌,从不含抑制剂的多孔状结构转变为含PVCap-XA1 的层层叠加“鱼鳞”状,可能后者结构更为致密,可阻碍甲烷气体进入水合物笼中。
猜你喜欢
石油化工技术与经济(2022年3期)2023-01-07 14:50:42
云南化工(2021年6期)2021-12-21 07:31:10
铜业工程(2021年1期)2021-04-23 01:45:06
西南石油大学学报(自然科学版)(2019年4期)2019-11-04 00:34:28
测控技术(2018年7期)2018-12-09 08:58:42
北方人(2018年16期)2018-08-20 06:01:22
科学与财富(2018年33期)2018-01-02 11:55:50
爱你(2016年4期)2016-12-06 05:15:27
石油化工技术与经济(2016年1期)2016-04-07 13:33:22
石油化工建设(2016年6期)2016-02-27 15:03:27
