
煤制乙炔关键中间体BaC2合成的热力学分析
2022-05-26 03:00:36李淼赵虹姜标陈思远闫龙
化工学报 2022年5期
李淼,赵虹,姜标,陈思远,闫龙
(1 中国科学院上海高等研究院,上海 201210; 2 中国科学院大学,北京 100049;3 榆林学院化学与化工学院,陕西榆林 719000)
引 言
煤炭从能源功能向资源功能转变是实现“碳中和”目标的最重要途径,发展绿色煤化工是践行“双碳”目标的重要举措。我国具有“富煤、贫油、少气”的能源结构特点,发展新型煤化工对于保障国家能源安全具有重要的战略意义。以煤为原料制备电石生产乙炔工艺在我国有70余年的历史,乙炔是我国生产聚氯乙烯(PVC)的关键原料,同时也是氯碱化工中平衡氯的重要手段,煤制电石乙炔在我国形成了成熟且规模巨大的乙炔煤化工生产链,在工业生产中占有重要地位[1]。由于强劲的市场需求,2020年电石产量达到了2792万吨,实现了产销量与价格的双增长[2-3]。
煤制电石乙炔的原理十分简单。首先由焦炭(C)和氧化钙(CaO)生成电石(CaC2)这一关键中间体,然后由电石与水反应放出乙炔。相比于煤制甲醇烯烃工艺,煤制电石乙炔工艺具有流程简单、建设周期短及投资少等优点[4]。并且1分子乙炔仅消耗1分子水,这样的低水耗大大降低了煤化工过程对水资源的依赖性。但另一方面,由于氧化钙与碳反应合成电石的热力学起始反应温度在1800℃以上,工业生产中采用电弧炉生产电石。为了强化化学反应和传热传质过程,通常在高达2000~2200℃的高温下发生液态反应合成电石,每吨工业电石的能耗约3340 kW·h。经相变形成的高密度大块电石需破碎后与水反应生产乙炔,该过程又造成了电石粉尘污染[5-6]。并且由于氢氧化钙溶解度小,电石发气后生成的氢氧化钙难以与焦炭中的灰分分离而循环利用,还造成了严重的电石废渣污染[7]。同时,在独立的石灰煅烧制备氧化钙过程中,每吨氧化钙不仅耗能高达988 kW·h,还副产二氧化碳约0.87 t[8]。在电石工业生产过程中,每吨工业电石的二氧化碳排放总量达3.7 t,电石渣(干基)排放达1.2 t。由此可见,以上原因环环相扣,其问题的根本点在于煤制电石反应动力学条件的优化不能克服热力学的限制,无法从根本上解决煤制电石高温高能耗等问题;碳化钙作为煤基乙炔的关键中间体,不能通过钙的化学形式的循环将原料中的C和水中的H转化为C2H2。为实现“30-60”碳中和目标,煤制电石生产工艺的零碳技术再造迫在眉睫。
同CaC2类似,BaC2作为一种离子型金属碳化物,其中碳以C2-2离子形式存在,与水反应时生成乙炔和氢氧化钡[9]。1892 年,将碳酸钡、碳、金属镁粉以一定的比例混合加热后,首次采用碳酸钡成功合成了碳化钡(BaCO3+3Mg+C===== BaC2+3MgO),该产物和水反应后生成乙炔气体;1894 年以氧化钡、碳酸钡和碳为原料,用电弧加热的方法再次合成了碳化钡。如表1所示,在随后的百余年中,有学者陆续对碳化钡的合成、晶体结构和性质等进行了研究。随着石油开采和冶炼技术的成熟,乙烯化工在世界范围内快速强势发展,而乙炔化工逐渐衰落。另一方面,由于煤制电石乙炔技术难度大,有限的研究也多集中在工艺条件的优化和电石炉型的改进。煤制碳化钡乙炔也始终没有得到足够的重视和产业界的关注。

表1 BaC2的研究发展历史Table 1 Research and development history of BaC2
我国是世界上最大的电石生产国和消费国,同时也是世界上钡资源最丰富的国家[28],重晶石资源储量和产量均居世界首位,如果采用碳化钡替代电石制乙炔具有丰富的原料来源。碳化钡的合成温度较碳化钙的低,且发气后形成的氢氧化钡在热水中的溶解度大,回收利用非常方便。另外,氢氧化钡与二氧化碳反应活性高,通过吸收外来二氧化碳又可以变为碳酸钡。所以,采用碳酸钡替代氧化钙,采用成型原料“一步法”固相合成碳化钡替代传统块状原料液相反应制备碳化钙,通过BaCO3-BaC2-Ba(OH)2-BaCO3的钡循环来进行煤制乙炔,有望在较为温和的条件下实现零碳排放和低固废排放的煤制乙炔,是一条绿色节能环保的煤制乙炔新路线。
热力学分析可以在很宽的温度和压力范围内,分析反应过程中可能存在的所有组分的固、液和气态情况,是研究化工过程、预测反应体系中反应物和产物变化的重要手段。本文将以热力学分析为主要手段,系统研究碳化钡合成的路径、可能存在的中间产物以及可能发生的副反应,预测较优的热力学合成条件、碳化钡的理论收率及获得高品质碳化钡的可操作弹性范围,并以碳酸钡和椰壳炭为原料进行了固相合成碳化钡的验证实验。研究结果可以为煤制碳化钡工艺优化提供参考依据,并为解决煤制电石生产过程中高碳排放、高能耗、高污染等问题提供新的思路。
1 BaC2合成反应体系的热力学分析
1.1 BaC2合成反应体系可能发生的反应
以BaCO3和C 为原料制备BaC2的体系中,可能会发生以下多种反应。
(1)从BaCO3出发可能发生的反应
BaCO3直接分解生成BaO:
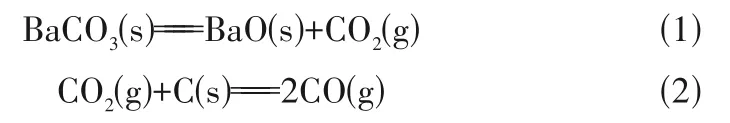
BaCO3和C反应生成BaO:
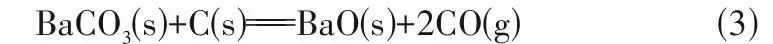
BaCO3不经过分解,直接和C 一步反应生成BaC2:

(2) 从BaCO3生成的中间产物BaO 出发可能发生的反应
BaO和C一步生成BaC2:

BaO 在C 的存在下,可能首先被还原成Ba(g)和CO(g)[13],然后Ba(g)和C生成BaC2:

(3)另外,碳化物在高温时,还可能发生分解的副反应,生成金属蒸气[11]。因此,在本反应体系中还可能存在BaC2分解的副反应。
首先,BaC2具有还原性,可能与中间产物BaO反应,生成Ba(g)和CO(g):
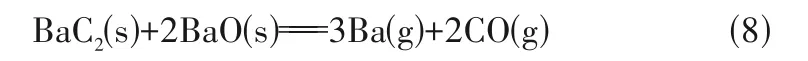
另外,BaC2在高温下还可能直接分解成Ba(g)和C:

1.2 热力学基本数据及计算方法
由于BaCO3、BaO 及BaC2的熔点分别为1740℃、1923℃、2000℃,在本文考察的温度范围内仅考虑这三种含钡的化合物的固态情况。因此,本考察体系中涉及的物质有C(s),BaCO3(s),BaO(s),BaC2(s),CO(g),CO2(g),Ba(g),其标准摩尔生成焓、标准熵、摩尔比定压热容来自于HSC-Chemistry 6.0 软件数据库,数值见表2,其中没有特别标注的物质状态均为固态。

表2 体系中物质的热力学数据(298.2 K)Table 2 Thermodynamic properties of substance used in present system(298.2 K)
根据式(11)~式(12)计算得到298 K 任意反应式(10)的标准摩尔反应焓及标准摩尔反应熵:
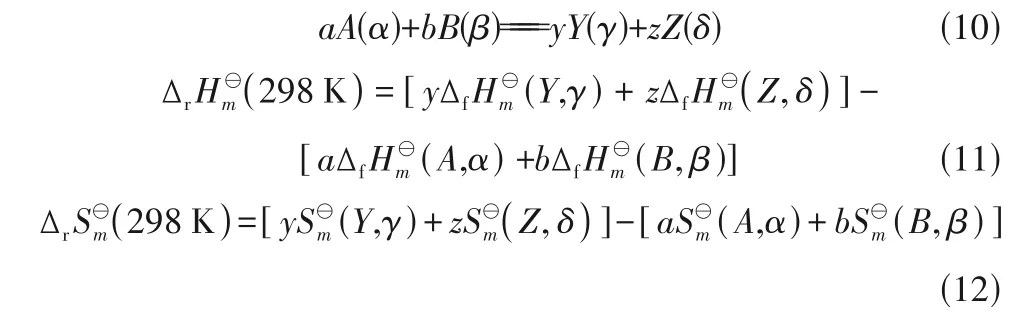
根据式(13)~式(16)计算得到任意温度下,反应的标准摩尔反应焓、标准摩尔反应熵、标准摩尔吉布斯自由能变。计算过程中气体按照理想气体,即各气体组分的逸度因子为1,根据式(17)可计算得到一定温度、压力条件下反应的摩尔吉布斯自由能变。

在恒温、恒压且非体积功为0的条件下,系统吉布斯函数减小的反应能自发进行,吉布斯函数不变,反应平衡,故判断反应能够自发进行的起始反应温度需满足的条件为ΔrGm= 0。当ΔrGm= 0 时lnK= -ΔrG⊖m/RT,由此可计算得到不同反应温度下的反应平衡常数。
通过分析比较各反应的起始反应温度的高低以及平衡常数的大小,可推测各反应发生的难易程度,从而可以推测热力学占优的BaC2的生成路径。同时还可以根据压力、温度对反应平衡常数的影响规律推测较为合适的动力学条件范围,并根据不同条件下各组分的平衡含量,初步确定获得高品质BaC2的操作区间。本文应用HSC-Chemistry 软件的平衡组成模块来计算体系的热力学平衡组成,应用吉布斯最小自由能法,在满足物料平衡方程的条件下,计算得到在指定的反应温度及压力下,使体系的吉布斯自由能达到最小值时的系统组成[29]。
2 实验材料及方法
2.1 实验试剂与原料
实验所用椰壳炭购买于艾格尼斯环境科技公司,使用前采用氢氟酸处理除去灰分杂质并在氩气气氛中900℃预处理3 h 除去可能的挥发分后,测得其固定碳含量为99.8%;所用的碳酸钡为分析纯,购买于上海泰坦科技有限公司;所用的氩气购于液化空气(上海)气体有限公司,纯度为99.9995%。
本实验采用湿法造粒制备反应原料小球。将粒度为0.15~0.18 mm 的椰壳炭和碳酸钡按照固定碳与碳酸钡的摩尔比为4∶1 混合均匀后,加入质量分数20%的去离子水后进行湿法造粒,制得直径为5 mm 的反应物小球,并置于120℃的烘箱中干燥12 h备用。
2.2 实验装置与实验过程
碳化钡合成的验证实验在高温固定床反应器中进行,以具有石墨内衬的刚玉管为反应管,其内径为40 mm,氩气为保护气,氩气流速为100 ml·min-1,装置示意图如图1 所示。实验时,考虑到刚玉管不能骤冷骤热的特性,首先以5℃·min-1的升温速率升至900℃,再以2℃·min-1的升温速率分别升至1050、1550℃,于1050、1550℃保温60 min。反应管后端联有在线气相色谱仪,可以对反应过程中的气相产物进行在线定量分析。

图1 合成BaC2反应装置Fig.1 Experimental setup of synthesis of BaC2 reaction
2.3 分析方法

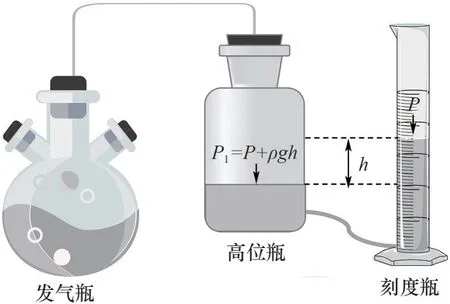
图2 产物发气测量装置Fig.2 Experimental setup of gas measuring
固体产物的物相分析在Rigaku MiniFlex 600 型X 射线衍射仪上进行,射线源为Cu 靶Kα射线,λ=0.154 nm,扫描范围2θ=10°~70°,扫描速率5(°)·min-1,管电压40 kV,管电流15 mA,步长0.02°。样品置于带有惰性气体保护的密封舱中进行XRD 检测。加热过程中的气相产物在SHIMADZU 2014C 型气相色谱仪上进行在线成分分析,检测器为热导检测器(TCD),色谱柱为Porapak Q 2 m×4 mm,载气为氩气,柱温70℃,柱流量2 ml·min-1。采用绝对校正因子定量法对气体产物进行定量分析,测得不同反应温度下尾气中一氧化碳、二氧化碳以及氢气的含量。其中一氧化碳、二氧化碳、氢气的绝对校正因子分别为fCO=8.91×10-7,fCO2=1.44×10-6,fH2=8.31×10-8。
3 结果与讨论
3.1 BaC2合成反应体系的主副反应分析
如前所述,本反应体系中可能发生的化学反应有9 种。表3 给出了反应温度在1273~2273 K 范围内各反应的标准摩尔反应焓。从表3 可以看出,在考察的温度范围内,除Ba(g)与C 合成BaC2[Ba(g)+2C(s)==BaC2(s)]的反应外,其他反应的标准摩尔反应焓均远大于零,说明这些反应均为强吸热反应。随着温度的升高,它们的标准摩尔反应焓呈减小的趋势,说明高温下这些反应的吸热量变小,高温有利于反应向正方向进行。
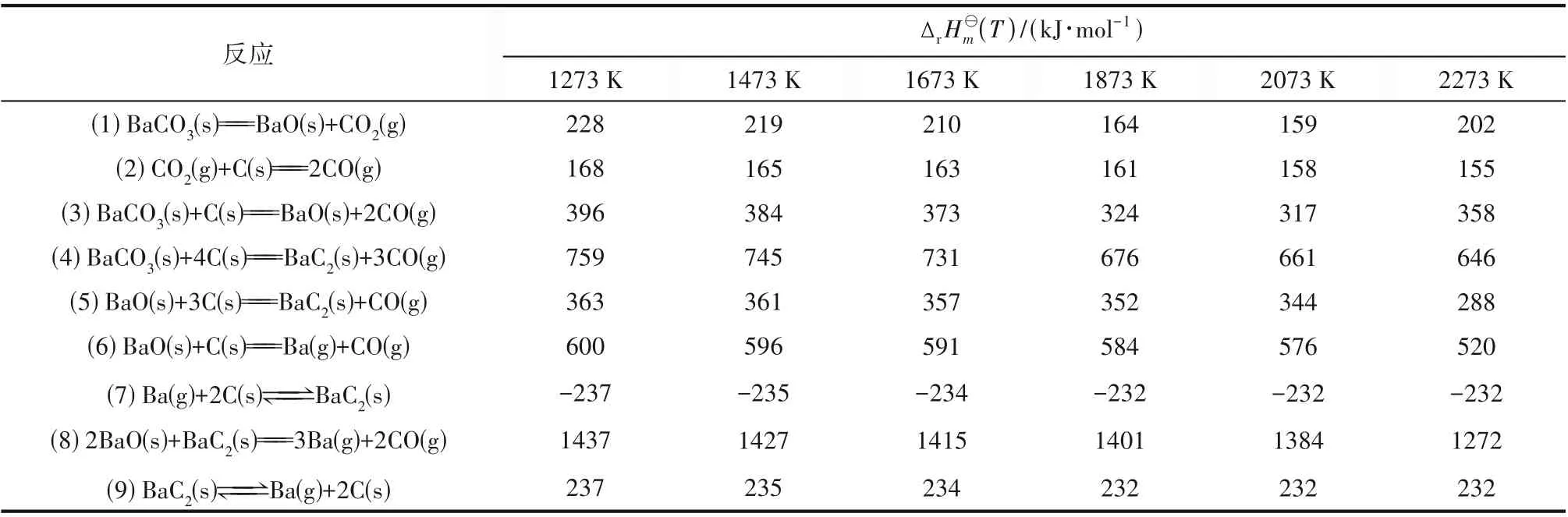
表3 不同温度下各个反应的标准摩尔反应焓Table 3 Standard molar reaction enthalpy change of reactions at different temperatures
表4 给出了常压条件下反应体系中各反应在1273~2273 K 温度范围内的标准摩尔吉布斯自由能变以及它们的热力学起始反应温度。从表4可以看出,除了Ba(g)与C 合成BaC2的反应式(7)外,其他各反应的标准摩尔吉布斯自由能变随着温度升高而降低,表明高温有利于绝大部分反应的进行。在表4 的反应中,仅有反应式(7)的ΔrGm在考察的温度范围内始终小于零,表明仅有Ba(g)与C 合成BaC2的反应在考察的温度区间一直是自发进行的。对其余8 个反应而言,CO2与C 反应生成CO 的Boudouard反应的起始反应温度最低,为973 K,表明以BaCO3与C为原料制备BaC2过程中,BaCO3分解的CO2很容易与C 转化为CO。特别需要指出的是,从表4 可以看出,从BaCO3出发的三个可能发生反应中,反应式(1)BaCO3分解为BaO和CO2的起始反应温度最高,为1831 K。反应式(4)BaCO3与C 直接生成BaC2和CO 的起始反应温度次高,为1515 K。反应式(3)BaCO3与C 生成BaO 和CO 的起始反应温度最低,为1320 K。这组数据表明从BaCO3出发的反应中,BaCO3与C生成BaO和CO的反应在热力学上占优。

表4 不同温度下各个反应的标准摩尔吉布斯自由能变(101 kPa)Table 4 Standard moore Gibbs free energy change of reactions at different temperatures(101 kPa)
图3(a)给出了从BaCO3出发的三个反应的反应平衡常数随温度的变化。如图3(a)所示,在反应物加热升温过程中,当温度上升到1320 K 后,BaCO3和C 最容易反应生成BaO 放出CO[反应式(3)BaCO3(s)+C(s)==BaO(s)+2CO(g)],且反应速度随着温度升高而加快。当温度升高到1515 K 时,反应式(4)BaCO3(s)+4C(s)==BaC2(s)+3CO(g)的lgK大于零,表明此时可能发生BaCO3与C 直接生成BaC2的反应,但当温度低于1805 K 时,它的反应平衡常数仍低于反应式(3)的反应平衡常数,表明当温度低于1805 K时,BaCO3与C 反应直接生成BaC2在热力学上并无优势。所以,可以认为反应物原料升温过程中首先生成了BaO中间体。仅有在快速的升温过程中,才可能存在BaCO3与C直接合成BaC2的情况。
从表4 还可以看出,从BaO 出发可能发生两个反应:反应式(5)BaO 与C 直接生成BaC2与CO,其起始反应温度为1806 K;反应式(6)BaO 与C 生成钡蒸气Ba(g)和CO,在1273~2273 K 温度范围内其吉布斯自由能变始终为正值,表明该反应在这个温度区间难以发生。图3(b)给出了这两个反应的反应平衡常数随温度的变化趋势。从图3(b)可以看出,随着温度的升高,两反应的反应平衡常数增大,但在相同温度下反应式(5)的反应平衡常数始终大于反应式(6),说明在所考察的温度范围内,反应式(5)BaO与C 直接生成BaC2与CO 的反应在热力学上更占优势。所以,从图3(a)、(b)可以看出,以BaCO3、C 为原料合成BaC2反应体系中,热力学上占优势的反应路径为BaCO3与C 先反应生成BaO 中间体,BaO 再与C一步反应生成产物BaC2。
由表4 和图3(c)还可知,反应体系中产物BaC2可能从两个路径发生分解副反应,反应式(9)BaC2直接分解为Ba(g)与C,反应式(8)BaC2与中间产物BaO反应生成Ba(g)与CO。反应式(8)、式(9)的吉布斯自由能变在1273~2273 K 范围内始终为正值,直到温度分别达到2448 K和3636 K以后才为负值,表明反应式(8)、式(9)在考察的温度范围内很难发生,也意味着产物BaC2的分解温度很高,远远高于其合成温度,在温度低于2273 K 时,BaC2难以分解。这一分析清楚地表明,与电石合成过程中碳化钙容易分解为钙蒸气导致钙流失不同[31-32],碳化钡合成体系中钡很难以钡蒸气的形式流失,这一特点有利于高品质碳化钡的生产。

图3 体系中可能发生反应的K-T曲线[(a)~(c)];CO分压对起始反应温度的影响(d)Fig.3 K-T curves of the possible reaction in the system[(a)—(c)]and influence of CO pressure on critical temperature(d)
由此可以推测:在以BaCO3和C 为原料合成BaC2时,热力学占优的合成反应路径为BaCO3先与C 反应生成CO 和BaO 中间体,然后BaO 继续与C 反应生成BaC2放出CO,即发生了BaCO3(s)+C(s)BaO(s)+2CO(g)、BaO(s)+3C(s)BaC2(s)+CO(g)这两步反应。该过程中无CO2的产生,副产物仅为CO。同时,相较于电石合成反应而言,合成BaC2的标准摩尔反应焓ΔrH⊖m为355 kJ·mol-1,小于合成CaC2的标准摩尔反应焓ΔrH⊖m458 kJ·mol-1,表明合成BaC2需要的能耗更少。
3.2 CO分压对反应体系的影响
在反应体系中,生成1 mol 的BaC2会放出3 mol的CO,因此CO 分压会严重影响化学反应平衡。图3(d)给出了CO 分压对BaC2合成反应和分解反应的影响。由图3(d)可知,随着CO 分压的降低,各反应的起始反应温度也随之降低。当CO 分压由0.1 MPa 降至0.01 MPa 时,从BaCO3出发可能发生的反应式(3)BaCO3(s)+C(s)BaO(s)+2CO(g)的起始反应温度从1320 K 降至1240 K,反应式(4) BaCO3(s)+4C(s)BaC2(s)+3CO(g)的起始反应温度从1515 K 降至1458 K。这表明降低CO 分压有利于BaO 及BaC2的生成,但BaCO3与C 反应生成BaO 的反应依然在热力学上占优。当CO 分压由0.1MPa 降至0.01 MPa时,反应式(5)BaO(s)+3C(s)BaC2(s)+CO(g)的起始反应温度由1806 K 降低至1646 K,反应式(6)BaO(s)+C(s)Ba(g)+CO(g)的起始反应温度由2275 K 降低至2112 K。这表明降低CO 的平衡分压同时降低了BaO 生成Ba(g)和生成BaC2两个反应的起始反应温度,但BaO 与C 反应生成BaC2和CO 这一反应依然占优势。并且,BaC2的起始分解温度虽然随着CO分压降低相应地降低,但仍远高于BaC2生成温度。因此,从以上分析可以看出,降低CO 分压可以降低BaC2的起始合成反应温度,但并不能改变BaC2生成的热力学占优的反应路径。在0.01~0.1 MPa的压力范围内,热力学占优的BaC2生成路径仍为BaCO3首先与C 反应生成BaO 中间体,然后BaO 与C 继续反应生成BaC2。即BaCO3(s)+C(s)BaO(s)+2CO(g),BaO(s)+3C(s)aC2(s)+CO(g)。
3.3 BaC2合成的热力学操作区间
图4 给出了反应物原料C/BaCO3的摩尔比为4时,反应系统各组分的平衡含量。图4(a)为CO 分压为0.1MPa时,各组分的平衡含量随温度的变化。由图4(a)可知,由BaCO3生成BaC2包含两个阶段,在1320~1373 K 范围内BaCO3与C 的物质的量减少,BaO 与CO 的物质的量增加,说明反应式(3)BaCO3(s)+C(s)==BaO(s)+2CO(g)将会先发生。在1373~1806 K范围内,各组分的物质的量不变,该温度范围内没有反应发生。在1806~1853 K 范围内BaO 与C的物质的量减少,BaC2产生,CO的物质的量增加,故该温度范围内将会发生反应式(5)BaO(s)+3C(s)==BaC2(s)+CO(g)。继续升高温度至2200 K,CO 的物质的量保持不变,BaC2的物质的量由1853 K 时的1 mol 降低至2200 K时的0.97 mol,同时体系中有0.03 mol的Ba(g)产生,说明常压条件下BaC2在高至2200 K的温度下都很难发生分解反应。
图4(b)给出了CO 分压降低至0.01 MPa 时,各组分平衡含量随温度的变化。由图4(b)可知CO 分压为0.01 MPa时,在1240~1283 K范围内BaCO3与C的物质的量减少,BaO 与CO 的物质的量增加,说明同样将会首先发生反应式(3)BaCO3(s)+C(s)===== BaO(s)+2CO(g),但BaCO3完全分解为BaO 的反应温度由1373 K 降低至1283 K。继续升高温度至1646 K,在1283~1646 K 范围内各组分的物质的量不变,没有反应发生。在1646~1696 K 范围内BaO 与C 的物质的量减少,BaC2、CO 的物质的量增加,在该温度范围内将会发生反应式(5)BaO(s)+3C(s)===== BaC2(s)+CO(g)。降低CO分压,BaO 与C完全反应生成BaC2的反应温度由1853 K 降低至1696 K。继续升高温度至2200 K,BaC2的平衡含量降低为0.81 mol,这表明在低CO分压的条件下,过高的温度会使BaC2分解。
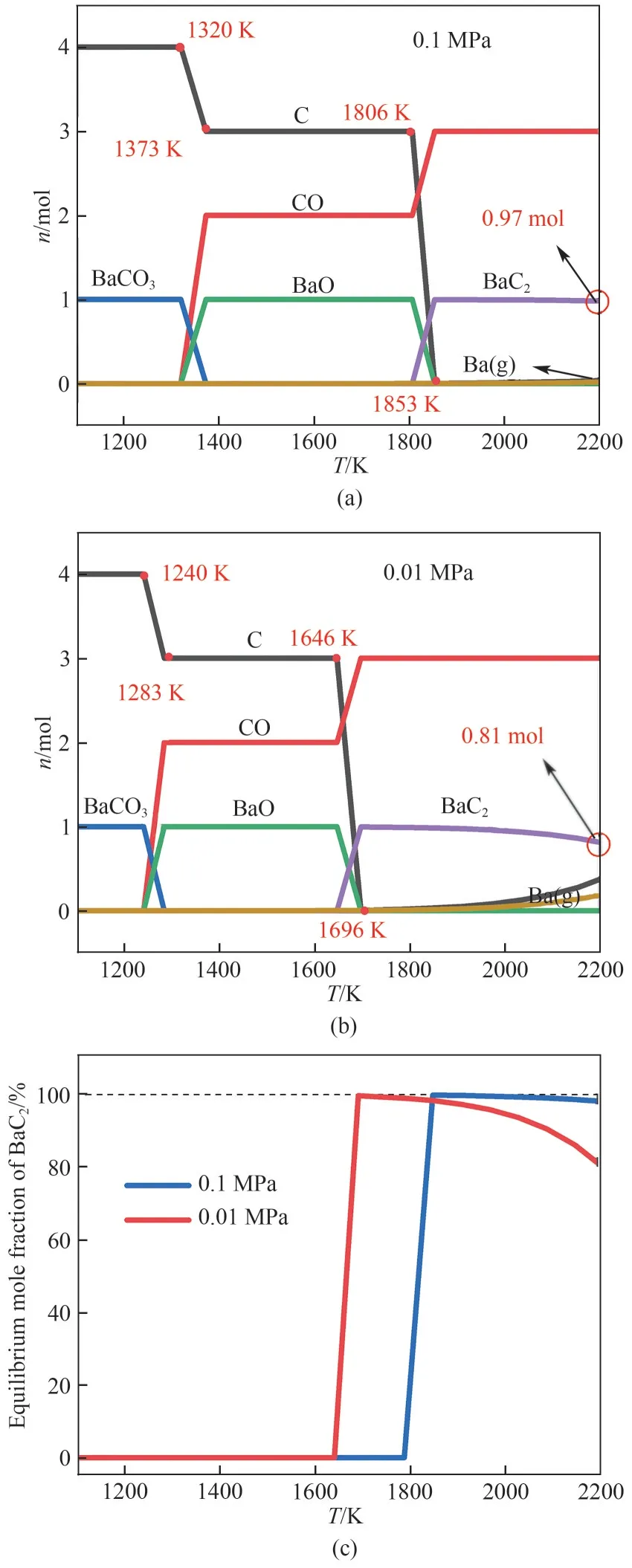
图4 不同温度下CO分压为0.1 MPa(a)、0.01 MPa(b)时的各组分含量以及BaC2的平衡含量(c)Fig.4 The content of each component under CO pressure of 0.1 MPa(a),0.01 MPa(b)and equilibrium mole fraction of BaC2(c)at different temperatures
图4(c)进一步给出了C/BaCO3摩尔比为4 时,BaC2的平衡摩尔分数随CO 分压及反应温度的变化。由图4(c)可知,当CO 分压为0.1 MPa 时,可在1853~2153 K 获得质量分数为99%以上的BaC2,当CO 分压降低到0.01 MPa 时,在1696~1853 K 的温度区间可以得到质量分数为98%以上的BaC2。由此可以看出,在较宽的温度和CO 分压范围内,都可以得到高含量的BaC2产物,满足工业生产的需求,操作弹性大。
3.4 BaC2合成的验证实验
为了验证热力学分析结果,采用固定碳含量为99.8%的椰壳炭为碳源,BaCO3为钡源进行了BaC2合成的验证实验。取20.3 g 充分混合好的反应物小球置于反应器中,程序升温到1823 K 并恒温加热1 h。加热过程采用在线气相色谱分析气相产物组成,加热结束后得到的固相产物进行XRD 分析和BaC2含量分析。
在加热过程中,气相色谱检测到反应尾气中含有CO 以及少量的H2,但未检测到CO2。图5(a)给出了尾气中CO 和H2的浓度随加热时间的变化。由图5(a)可知,加热140 min 后,反应温度达到973 K,此时出口气中检测到了少量的CO,其浓度为0.4 ml·min-1。随着温度升高,CO 浓度增加,当反应温度升高到1073 K 时,反应尾气中除了检测到浓度为4.1 ml·min-1的CO 以 外,还 检 测 到 了 浓 度 为0.4 ml·min-1的H2。 随着温度升高,CO 和H2的浓度上升,当温度升高至1223 K,尾气中H2的浓度达到最大值5.0 ml·min-1,而CO浓度继续上升,直到温度升高到1323 K 时,CO 浓度达到第一个峰值40.6 ml·min-1,但此时氢气的浓度降低到了0.9 ml·min-1。随后继续升温,尾气中CO 浓度由40.6 ml·min-1迅速降低,H2的浓度由0.9 ml·min-1继续降低。当反应温度升高至1423 K 时,反应尾气中检测不到H2。尾气中少量的H2可能是来自于反应物中少量的水分或者羟基基团与碳发生的反应H2O+CH2(g)+CO(g)。当温度升高到1473 K 时,CO 的浓度降低到第一个最低值2.9 ml·min-1。随着加热时间的延长,反应温度继续升高,尾气中CO 的浓度又开始升高,当加热时间为505 min,反应温度达到1823 K时,CO浓度达到第二个峰值,为16.0 ml·min-1。此后将反应物料在1823 K 恒温,发现此时尾气中CO 的浓度开始降低,当保温时间延长至1 h,加热时间为565 min时,尾气中的CO 浓度降低到了0.6 ml·min-1,表明此时反应速率很慢,反应基本结束。由图5(a)可看出,CO 的生成经历了两个不同的反应阶段。根据前面的热力学分析,可以推断,这两个阶段应分别对应于反应式(3)BaCO3(s)+C(s)===== BaO(s)+2CO(g)和反应式(5)BaO(s)+3C(s)===== BaC2(s)+CO(g)。将CO 的瞬时浓度对加热时间进行积分计算,并将低温阶段可能与H2伴随产生的CO 扣除后,可以得到两个阶段产生的CO的总量,分别为3327 ml 和1531 ml,与20.3 g 原料在两个阶段应该生成的CO 理论值3667 ml 和1833 ml非常接近。这一结果表明了热力学预测的反应路径符合反应的真实情况。
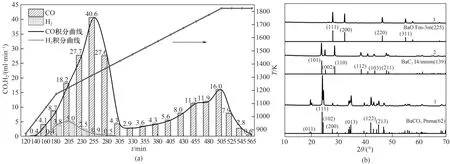
图5 反应产生CO的流量(a);反应物原料(1)以及在1823 K(2)、1323 K(3)下的反应产物的XRD谱图(b)Fig.5 The flow of CO produced by the reaction(a);XRD patterns of reactant(1)and products at 1823 K(2)and 1323 K(3)(b)
图5(b)给出了反应物原料以及不同温度下加热产物的XRD 谱图。图5(b)曲线1 为反应物原料的XRD 谱图,可以看出,23.9°、24.3°与27.1°处的衍射峰为BaCO3的特征峰。椰壳炭的石墨化程度较低,衍射峰不明显。图5(b)曲线2 给出了反应物原料在1823 K 加热1 h 后的产物的XRD 谱图。图中BaCO3的特征峰完全消失,同时在23.8°、25.2°与28.6°等位置出现了强度不一的衍射峰,为I4/mmm 晶型的BaC2的特征峰,且无明显的BaO 特征峰,表明此时BaCO3基本上转化为BaC2。将产物与水反应发气,并对发气后的溶液进行ICP 分析,可以得出BaCO3的转化率为100%,BaC2的选择性为96.2%,BaO 的选择性为3.8%,乙炔的收率为96.2%。图5(b)曲线3给出了反应物原料在1323 K 下加热1 h 后所得产物的XRD 谱图。可以看出BaCO3的特征峰完全消失,并在27.9°、32.3°与46.2°处出现了Fm-3m 晶型的BaO 特征衍射峰,同时图中没有观察到明显的BaC2特征峰,表明当温度加热到1323 K 时产物为BaO,BaC2难以生成。这一结果也验证了热力学分析的结果。
3.5 钡的循环
图6给出了Ca(OH)2、Ba(OH)2在水中的溶解度随温度的变化曲线。由图6可知,当温度由273 K升至353 K 时,Ca(OH)2的溶解度由0.185 g/100 g 水降至0.094 g/100 g 水,而Ba(OH)2的溶解度由1.670 g/100 g水升至100 g/100 g 水。在煤制乙炔过程中,CaC2、BaC2与水反应放出乙炔,同时生成Ca(OH)2、Ba(OH)2。在传统电石乙炔工艺中,由于Ca(OH)2溶解度小,且随着温度变化差异不大,难以与碳源带进的Si、Al、Fe、S、O 等元素组成的不溶性杂质分离而回收利用,从而产生了大量的电石渣。在新工艺中,BaC2与水反应生成乙炔为放热过程,副产的Ba(OH)2溶解在水中,与不溶性杂质通过热过滤很容易分离,从而实现Ba(OH)2的回收。在Ba(OH)2热溶液中通入CO2,可以容易地将Ba(OH)2转化为BaCO3,实现钡的循环利用。考虑到BaC2合成及发气过程中钡的质量平衡情况,采用ICP 法分析了产物发气后溶液中的钡含量,并与反应物原料小球中的钡含量进行了比较。20.3 g 反应物小球中Ba 含量为83.8 mmol,反应后将碳化钡产物与水反应发气,测得发气后的溶液中Ba 含量为82.5 mmol,由此可以算出钡的回收率为98.4%。其中损失的1.6%的钡是由于高温下反应物中的少量钡与石墨内衬发生了反应造成的。因此,当以BaC2为煤制乙炔的中间体时,钡元素通过BaCO3-BaC2-Ba(OH)2-BaCO3的循环,将C 和CO2转化为C2H2和CO。

图6 Ca(OH)2及Ba(OH)2在水中的溶解度[33]Fig.6 The solubility of Ca(OH)2 and Ba(OH)2 in water[33]
4 结 论
(1)用热力学计算的方法分析了BaC2合成反应体系中可能发生的各反应的热力学特点,推断了BaCO3首先与C 反应生成BaO 中间体和CO,然后BaO 与C 一步反应生成BaC2和CO 为热力学上占优的BaC2合成路径;考察了反应温度、CO 分压对BaC2合成反应以及各组分平衡组成的影响,当CO 分压由0.1 MPa降低至0.01 MPa时,BaC2合成的起始反应温度由1806 K 降低至1646 K,但没有改变热力学占优的BaC2合成路径;BaC2热稳定好,难以在高温下发生反应生成钡蒸气;在温度区间为1696~1853 K,压力区间为0.1~0.01 MPa,BaC2理论收率均可以达到98%以上,满足工业生产的需求。
(2)采用椰壳碳和碳酸钡原料体系对热力学分析结果进行了验证。反应体系在加热升温过程中没有检测到CO2产生,且CO 浓度随着加热温度升高呈现出显著的两步反应的特点;在1823 K 反应1 h后,产物中BaC2的质量含量为96.2%。实验结果很好地验证了热力学分析对碳化钡合成路径和反应条件预测的结论。
(3) 采 用BaC2替 代CaC2,通 过BaCO3-BaC2-Ba(OH)2-BaCO3的钡循环可以将C 和CO2转化为C2H2和CO。与电石路线相比,此路线反应温度更低,不仅无CO2排放,还可吸收CO2转化为CO,同时固废排放极大地减少,可为我国现代煤化工的绿色可持续发展提供新的思路。
符 号 说 明
Cp,m——摩尔比定压热容,J·mol-1·K-1
ΔrCp,m——反应前后物质的摩尔比定压热容差,J·mol-1·K-1
G——单位质量产物的发气量,ml·g-1
ΔrGm——任意温度任意压力下的摩尔吉布斯自由能变,kJ·mol-1
g——重力常数,9.8 N·kg-1
h——高位瓶与刻度瓶的液面差,m
K——反应平衡常数
m——用于发气的产物的质量,g
P——大气压,kPa
P′——在t℃时饱和食盐水蒸气压力,kPa
Pi——组分i的分压,kPa
R——摩尔气体常数,8.314 J·mol-1·K-1
Δr——标准摩尔反应熵,J·mol-1·K-1
T——温度,K
t——发完气后体系的温度,℃
V′——发气后由于高位瓶液面与刻度平液面的液位差,造成高位瓶内气压大于大气压力所引起气体体积减小的校正值,ml
ΔV——产物发气量,ml
vi——组分i的化学计量系数
ρ——饱和⊖食盐水的密度,kg·m-3
上角标
⊖——标准态
下角标
f——生成态
m——体系中的各物质
p——恒定压力
r——反应态
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:42
云南化工(2021年8期)2021-12-21 06:37:38
中国石化(2021年8期)2021-11-05 07:00:16
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21 07:48:02
上海节能(2020年3期)2020-04-13 13:16:12
中国盐业(2018年12期)2018-09-21 07:14:08
当代化工研究(2016年6期)2016-03-20 16:21:43
中国氯碱(2014年8期)2014-02-28 01:04:46
中国医药科学(2013年16期)2013-12-20 11:34:34
合成纤维工业(2013年6期)2013-12-08 07:26:48
