
非水相金属基离子液体湿法氧化脱硫工艺:发展与展望
2022-05-26 02:57白文轩陈锦湘刘芬张静淙谷志平熊成铭施王军余江
化工学报 2022年5期
白文轩,陈锦湘,刘芬,张静淙,谷志平,熊成铭,施王军,余江
(北京化工大学化工学院能源环境催化北京市重点实验室,北京 100029)
引 言
硫化氢广泛存在于化石能源的开采以及化工领域各个工业的生产过程,关系到国民经济发展建设的关键,如石油开采工段的油田伴生气[1-2]、炼油过程的炼厂干气及酸性水汽提尾气[3],克劳斯硫磺回收工艺段的加氢尾气[4],天然气和生物质燃气(沼气)的净化[5-6],精细化工业的废水经生化处理后的末端尾气等[7]。然而硫化氢的存在不仅严重危害了大气环境及人体健康[8-9],还会引起设备腐蚀以及催化剂硫中毒的现象[10-11]。此外,硫化氢的硫资源可以作为硫磺得到回收,带来巨大的经济效益。例如,作为国内储量最大的整装海相气田的普光气田,硫化氢平均含量高达15%,年处理天然气能力达120×108m3,年产硫磺高达200×104t[6]。因此亟需开发适用范围广且绿色化的原位净化工艺。尽管当前碱性水相湿法氧化脱硫工艺得到较为广泛的应用,但存在二次污染严重等诸多缺陷,由此借鉴离子液体新材料特点所发展的非水相脱硫工艺得到广泛关注。迄今为止,该工艺已经陆续开展了小试、中试以及示范应用研究[12]。本文将对新工艺发展历程以及离子液体脱硫体系结构特点、性能表征、脱硫工艺构建及关键科学问题进行综述。
1 离子液体湿法氧化脱硫技术的发展机遇
1.1 传统水相湿式氧化脱硫的局限性
传统水相湿式氧化法主要包括改良ADA 法、HPF法、Lo-Cat法。其中ADA 法和Lo-Cat均是利用Na2CO3溶液作为碱源来吸收H2S,而HPF 法则直接利用焦炉煤气的氨作为碱源,无须外加碱液,但存在氨气逸出的问题。改良ADA 法以V2O5为催化剂,蒽醌二磺酸钠(ADA)为氧载体,同时加入酒石酸钾钠来防止生成V-O-S沉淀[13]。HPF法以双核酞菁钴(PDS)为催化剂,脱硫过程中,H2S 首先被氨水吸收,形成了NH4HS和(NH4)2Sx,在再生过程中,HS-和S2-在PDS 催化剂上与O2发生电子转移,实现了硫化物的转化[14]。Lo-Cat 法是一种以螯合铁(NaFeEDTA)为催化剂的湿式氧化脱硫技术,该法利用三价铁与硫化氢的氧化还原反应来实现硫化氢的氧化,再通过氧气对亚铁离子进行再生。与ADA和HPF法不同,Lo-Cat 工艺在吸收阶段就产生了硫磺,而另外两种工艺则是在再生工段通过氧气的催化氧化来实现硫化物的转化[15-16]。
上述水相湿式氧化脱硫技术,存在反应控制与过程控制两大类难题。
反应控制:碱性脱硫体系极易受到原料气中酸性气体CO2的影响,如天然气中CO2浓度可达8%[17],生物沼气中CO2浓度更是高达15%~60%[5],因此需补充碱液调控pH(8~9);脱硫液的活性组分溶解性差,活性成分浓度低,导致硫容较低;脱硫富液氧化再生过程中,金属活性成分氧化再生效率低,滞后脱硫效率,影响硫磺品质[18];活性氧物种使得硫化氢过度氧化成副盐,造成二次污染。
过程控制:硫磺颗粒的形貌及成长,关系到脱硫体系的硫磺分离效率,需要具有表面活性的硫磺改进剂等辅助[19];再生过程的外场强化,如射流强化以实现空气中氧气在脱硫液中的增溶而提高再生效率[20],但不可避免地会产生活性氧物种的生成及过度氧化的难题,而氧化能力过强会导致脱硫剂降解[21],生成大量副盐从而抑制脱硫反应,同时影响硫磺纯度,产生高盐有机废水。
表1 综合了三种水相湿法氧化脱硫工艺特性,表明传统工艺无法从原理上突破技术瓶颈。

表1 三种水相湿式氧化脱硫工艺Table 1 Three types of aqueous wet oxidation desulfurization processes
1.2 非水相湿法氧化脱硫的机遇
天然气及油田伴生气等能源气的分布表现出地域位置偏、气井分散广、含硫浓度及天然气存量差异大的特点,并不适合对环境影响大的管道运输集中处理方式[30-31]。继承水相湿法氧化脱硫的脱硫效率高、适用范围广的优点,开发绿色无二次污染的小型撬装化工艺及装置,对满足上游产业的开采-净化-压缩储存-移动运输以及下游生产工艺的稳定运行具有极其重要的现实意义。
1.2.1 非水相湿法氧化脱硫体系的由来 水相湿式氧化脱硫存在药剂消耗量大和排放难处理有机废水等问题,不符合小型撬装化绿色脱硫工艺发展的需求。2006 年,Hua 等[32]报道了将FeCl3溶于NMP溶液中的一种非水相脱硫体系,脱硫效率可达99.72%,并且无须调控pH,脱硫过程不存在明显的脱硫液降解,基本无副盐产生。可见,非水相湿式氧化脱硫体系对解决水相湿法氧化脱硫的瓶颈难题有很好的借鉴作用。但该体系中三氯化铁含量低,需要添加少量H2O以增加脱硫液中的含铁量,提高脱硫容量。然而由水引发的脱硫剂降解、低沸点溶剂及其热稳定性差等难题仍难以克服,成为非水相湿法氧化体系发展的关键问题。
1.2.2 金属基离子液体的设计与合成 离子液体是由阴阳离子构成的离子型化合物,且在室温条件下是不易挥发的液体,其阴阳离子既可是无机离子也可是有机离子,表现出突出的可设计性、超溶解性、宽电化学窗口和高热化学稳定性的特点,作为一类新材料在分离和催化氧化领域得到广泛关注和应用[33-34]。Wu等[35]和马云倩等[36]利用1,1,3,3-四甲基胍(TMG)阳离子分别实现对H2S与SO2的高效吸收。咪唑基离子液体对不同种类的气体(CO2,N2,SO2,H2S,O2)均 展 现 出 了 较 强 的 吸 收 能 力[37],Sanchora 等[38]通过电子结构计算和AIM 分析研究了一系列[C2mim]+与不同阴离子合成的离子液体对H2S、SO2和CO2的相互作用,发现气体分子更容易与阳离子相互作用。此外,对于[Cnmim]+阳离子而言,烷基链的长度明显地影响了其对气体的吸收能力[39]。同样,阴离子对气体吸收也存在显著作用。金属卤化物型阴离子[MCln]-就容易与H2S 和NH3这类极性气体分子形成氢键,从而促进吸收[40];对于螯合型金属基离子液体,中心金属离子与气体分子间存在的配位作用有利于气体分子的吸收,比如希夫碱基离子液体对氧气分子就具有很强的吸收能力[41]。因此,将上述阴阳离子组合可以得到不同种类的金属基离子液体(MBILs)。
研究表明,氯化铝基离子液体的结构及酸性随摩尔比M[n(AlCl3):n(Emim)]的变化而发生变化:M<0.5 时,离子液体为碱性,阴离子主要为Cl-;M=0.5时,体系为中性,阴离子主要为AlCl4-;当M>0.5时,体系呈现出很强的Lewis 酸性,阴离子主要以Al2Cl7-和的形式存在,见图1[42]。2004年,Hayashi等[43]在开放的大气环境中用BmimCl 与FeCl3·6H2O 合成出了一种稳定的磁性铁基离子液体(BmimFeCl4),其结构变化与无水条件下合成的氯化铝基离子液体十分相似[42]。这种离子液体集[Bmim]+的稳定性与卤化金属盐如[FeCl4]-的催化氧化能力于一体,在催化分离领域具有很好的应用潜能。
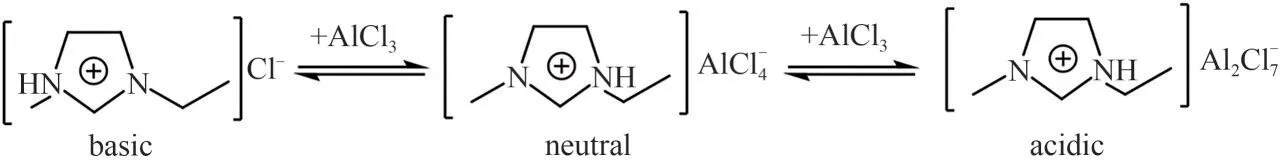
图1 AlCl3合成配比对铝基离子液体酸度及结构的影响[42]Fig.1 The effect of molar ratio of AlCl3 on the acidity and structure of aluminum-based ionic liquid[42]
因此,通过对金属基离子液体阴阳离子结构的设计,可定向合成同时具备吸收中心与催化中心的双活性位点的功能型离子液体[35,42]。图2 展示部分典型的作为气体吸收中心的有机阳离子与作为催化氧化中心的金属络合阴离子。除此之外,还可以有络合金属阳离子与有机阴离子组合的金属基离子液体(MBILs),有待进一步研究。

图2 四类典型的阴阳离子[44-48]Fig.2 Four types of typical anions and cations[44-48]
1.2.3 金属基离子液体氧化脱硫原理 何义等[49]于2010 年提出以铁基离子液体为脱硫剂的非水相湿式氧化脱硫(Nasil)工艺。铁基离子液体由BmimCl与FeCl3·6H2O 按照1∶2 的摩尔比合成所得,表现出Brönsted酸和Lewis酸的特点[50],具备很好的氧化性、氧化还原可逆性和热稳定性。如图3 所示,氯化咪唑铁基离子液体(BmimFeCl4)具有高的氧化还原电位(ORP)值,循环伏安曲线(CV)具有很好的对称性,表明铁基离子液体具有良好的氧化能力,且表现出良好的氧化还原可逆性。能够直接将H2S氧化为硫磺,过滤后得到亮黄色产物,氧化脱硫后的铁基离子液体可以被空气中的氧气氧化再生以循环使用。不仅如此,热重曲线(TG)表明其热分解温度达到350℃,变温拉曼光谱(VT-Raman)显示低于200℃的高温环境中,拉曼谱线没有变化,液体黏度没有增大,同时其CV 曲线仍然保持高度对称性,说明高温环境中铁基离子液体仍保持良好的氧化还原可逆性,没有发生变质,满足130~150℃直接熔硫的需要。

图3 BmimFeCl4的物化性能及氧化脱硫反应原理[49-50]Fig.3 The physicochemical properties and oxidative desulfurization reaction principle of BmimFeCl4[49-50]
郭智慧等[51]的研究表明,随着FeCl3·6H2O 用量的增加,产物BmimFeCl4的黏度明显降低;而使用无水FeCl3合成的Fe-IL,虽然红外光谱反映出的结构一致,但黏度明显偏高。马云倩等[52]将Et3NHCl与无水FeCl3按照1.6∶1 的摩尔比,在80℃下合成了1.6Et3NHCl·FeCl3离子液体,具备良好的热稳定性,但其黏度较大,达到457 mPa·s。
离子液体的超溶解性能够实现铁离子在有机溶剂中的增溶,明显地提升了体系的催化氧化能力。在非水相脱硫体系FeCl3/NMP 中,FeCl3浓度仅为90 mmol/L[32];而BmimFeCl4和Et3NHFeCl4中 的Fe(Ⅲ)浓度分别为3.55 mol/L 和3.41 mol/L,显著增加了液相中铁离子的含量,并且这两类离子液体能够与DMAC 和DMF 等有机溶剂互溶[49,52-53],解决了非水相体系中金属离子在有机溶剂中含量低的难题。由此,金属基离子液体的硫容得到显著提升,Et3NHFeCl4的静态硫容达到6.36 g/L[52],基于咪唑铁基离子液的Nasil 工艺动态硫容尽管常温时仅有0.31 g/L[49],但升高温度硫容随之增大,与非质子溶剂NHD复配后在1.7 MPa下硫容高达21 g/L以上[54]。
Nasil工艺脱硫机理与Lo-Cat法相同,均通过脱硫液中三价铁与硫化氢发生氧化还原反应实现硫化氢的转化,脱硫富液通过曝气的方式即可再生,产出的硫磺在熔硫釜内分离纯化(图3)。Nasil工艺硫容高,工艺流程简单,产品硫磺的品质较好,且无二次污染[33,55-56]。区别于水相氧化脱硫工艺,非水相工艺中再生曝气时氧气难以被活化,避免了硫化氢被过度氧化成和等副产物,因此分离出的硫磺纯度高达99.3%以上,无二次污染[57];此外,酸性体系脱硫过程中不易受到CO2的影响,因此不存在Lo-Cat 工艺中控制pH 的问题[53];咪唑阳离子的热化学稳定性较强,脱硫再生过程中不易被氧化降解,能耐受150℃以下的高温熔硫,脱硫液可循环使用[50]。由此可知,金属基离子液体氧化脱硫工艺能够突破传统水相氧化脱硫工艺的技术瓶颈,符合小型撬装化工艺设备的设计需求。
2 金属基离子液体湿法氧化脱硫工艺的发展
尽管以铁基离子液体为活性成分的氧化脱硫性能得到小试验证,但是BmimFeCl4和Et3NFeCl4存在黏度大传质效率低、易起泡等难题,对金属基离子液体湿法氧化脱硫工艺的放大应用十分不利。
2.1 氧化脱硫反应与外场强化
2.1.1 溶剂对铁基离子液体脱硫过程的影响 对于湿法催化氧化脱硫技术,溶剂对脱硫效率起着决定性的影响作用。研究表明,BmimFeCl4能与多数有机溶剂互溶,复配低黏度的有机溶剂明显降低铁基离子液体脱硫剂的黏度,促进硫化氢的吸收。被广泛用于提升铁基离子液体脱硫性能的溶剂主要包括聚乙二醇二甲醚(NHD)、N,N-二甲基乙酰胺(DMAC)、N,N-二甲基甲酰胺(DMF)、1,3-二甲基-2-咪唑啉酮(DMI)以及N-甲基吡咯烷酮(NMP)[55,58-60]。余江等[53]将BmimFeCl4与N,N-二甲基乙酰胺(DMAC)按照9∶1 的质量比配制脱硫剂,在293 K 下,其黏度由30.8 mPa·s 下降至8.67 mPa·s,气液传质性能也大幅度提升。BmimFeCl4中[Bmim]+阳离子与[Fe(Ⅲ)Cl4]-阴离子组成的离子对,随着有机溶剂的添加,循环伏安曲线结果表明阴阳离子先由接触离子对形态(CIP)逐步拉开间距演变成双溶剂分离离子对(2SIP)的形态[53,61],见图4(a)。复配NHD 有机溶剂后,消弱了[FeCl4]-与咪唑阳离子之间的作用力,导致体系的离子扩散速率和电导率提升,同时黏度也会相应降低。NHD 的氢键作用促进硫化氢分子溶解在Fe-IL/NHD 脱硫体系当中,首先被解离成H+和HS-,再被Fe-IL 中[Fe(Ⅲ)Cl4]-氧化为硫单质,而[Fe(Ⅲ)Cl4]-被还原为[Fe(Ⅱ)3Cl7]-,见图4(b)[55,58,62]。
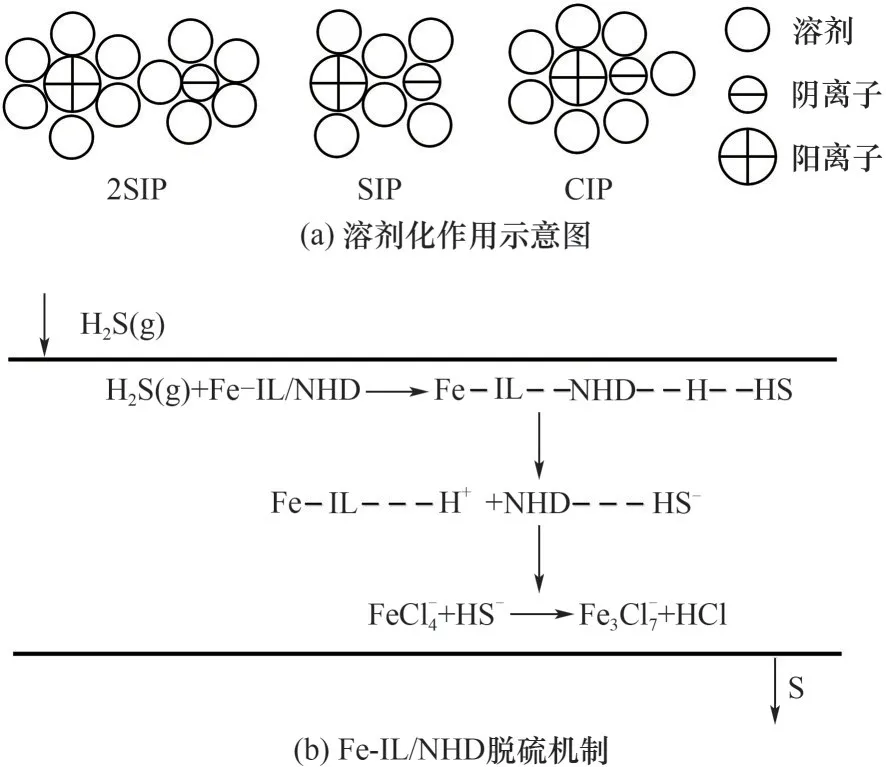
图4 溶剂化作用示意图以及铁基离子液体共溶体系(Fe-IL/NHD)脱硫机理[58,61]Fig.4 Schematic diagram of solvation and desulfurization mechanism of iron-based ionic liquid compound system(Fe-IL/NHD)[58,61]
对比复配NHD、NMP、DMI、DMF 这四种溶剂后的铁基离子液体Fe-IL 脱硫性能发现,Fe-IL/NHD的脱硫效率明显弱于另外三类溶剂,Fe-IL/NHD 和Fe-IL/NMP 脱硫体系中Fe的反应级数分别为1级和2 级[59,62],说明溶剂类型对铁离子活性存在着极大的影响。阴离子和阳离子倾向于在低介电常数的溶剂中形成接触离子对,在高介电常数的溶剂中则容易被分离[63-64],除此之外,氢键对离子液体的溶剂化作用也具有显著影响[65-66]。Zhang等[67]通过DFT计算证明甲醇分子能够通过π-π 堆叠和氢键的共同作用影响BmimFeCl4的分子结构形态。NMP 分子的强极性既能够强化铁基离子液体的溶剂化效应,又能提供大量氢键供体和受体以活化[FeCl4]-中的Fe3+与HS-中的S2-,从而提升脱硫体系的转化率。
2.1.2 水对铁基离子液体脱硫过程的影响 虽然BmimFeCl4本身是疏水体系,但复配溶剂的加入改变了其亲疏水性能。而铁基离子液体脱硫系统中的水主要来自原料气与空气氧化的再生过程[68]。从物化特性上看,水会对体系的黏度和电导率造成显著的影响,少量水(<10%)的存在下,离子液体的黏度也会发生明显的降低,从而更有利于气液传质过程[69-70]。
Hua 等[32]研究了水对Fe3+/NMP 体系脱硫的影响,加入5%的水,可能会产生氧化活性较高的中间产物Fe(OH)2+,从而推动反应的进行。Ebrahimi 等[71]对脱硫反应动力学研究结果与Hua 等观点一致,见式(1)~式(3),其中式(2)为不可逆二级反应,是总反应[式(1)]的决速步骤。铁基离子液体虽为非水相脱硫体系,但水分子与离子间的相互作用对Fe(Ⅲ)的还原也有一定的影响。根据Wang 等[72-73]的研究,少量水(<10%)的引入,能够破坏阴阳离子间的相互作用,水分子既能够与咪唑环形成氢键[74],又能够与亲水阴离子(Cl-)作用[72],弱化Fe-Cl 的键合强度,提升脱硫性能。

2.1.3 多金属离子协同作用对铁基离子液体脱硫过程的影响 向BmimFeCl4引入其他金属离子,如Fe(Ⅱ)、Cu(Ⅱ)、Mn(Ⅱ)、Zn(Ⅱ)等,将改变Fe-IL 的阴阳离子空间结构以及脱硫过程的反应机理[58,60,75-76]。然而这四种金属基离子液体([Bmim]2[M(Ⅱ)Cl4])的阴阳离子结合能明显高于BmimFeCl4[77],这使得它们的黏度较大,降低了脱硫体系的传质性能。
严召等[76]使用Zn/Fe 体系的水相湿法催化氧化脱除沼气中的H2S,指出Zn2+作为溶液中S2-的捕捉剂,通过生成ZnS 沉淀的方式促进H2S 的氧化。余江等[68,75]报道了Fe/Cu 复合离子液体脱硫体系的脱硫效果,发现Cu2+能够提升脱硫效率。但非水相金属基离子液体的酸度能使CuS 和ZnS 溶解,碱性水相体系和酸性非水相体系的反应机理是有区别的,其中氧化性的Cu(Ⅱ)与H2S 快速反应生成CuS,继续与氧化性Fe(Ⅲ)反应而转化为S 和还原性的Fe(Ⅱ),而Cu(Ⅱ)在将还原态Fe(Ⅱ)氧化为Fe(Ⅲ)的同时自身转化为还原态Cu(Ⅰ),而还原性的Cu(Ⅰ)易被O2氧化,恢复为Cu(Ⅱ),由此构成一个循环。反应中Cu(Ⅱ)能优先捕获H2S,为发挥Fe(Ⅲ)的氧化作用提供了前体(图5)。
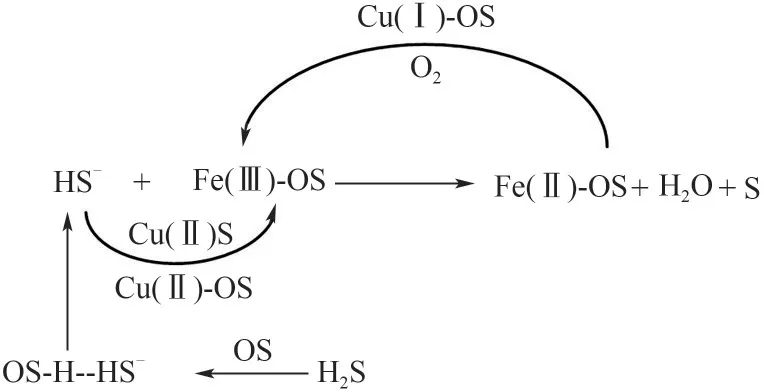
图5 二元金属离子对脱硫过程的协同强化机制OS—有机溶剂;Fe(Ⅱ)—二价铁离子;Fe(Ⅲ)—三价铁离子;Cu(Ⅱ)—二价铜离子;Cu(Ⅰ)—一价铜离子Fig.5 Synergistic mechanism of binary metal ions on desulfurization processOS—organic solvent;Fe(Ⅱ)—ferrous ion;Fe(Ⅲ)—ferric ion;Cu(Ⅱ)—copper ion;Cu(Ⅰ)—cuprous ion
Guo 等[58]对Fe(Ⅲ/Ⅱ)-IL 脱硫体系的研究表明,Fe(Ⅲ/Ⅱ)-IL 在270 min 时的脱硫效率仍大于90%,而Fe(Ⅲ)-IL 在同等条件下脱硫50 min 后,脱硫效率明显降低,因此二价铁引入有助于提高Fe(Ⅲ)的利用效率,使得S2-更容易被氧化。胡锦超等[60]的研究表明,对于二元脱硫体系Fe/Mn-IL 和Fe/Zn-IL,前者脱硫能力明显优于后者,但是同时复配了DMI后,后者的脱硫性能却更为优越。复合金属离子之间会形成M-Cl 的竞争配位,因此弱化了Fe-Cl 键的强度。引入有机溶剂后,溶剂化作用在一定程度上增大了离子间距,弱化了多金属离子与氯离子之间的竞争配位作用。此时多中间价态的金属离子作为氧化还原介体可对脱硫反应过程进行强化[68]。
2.1.4 B-L酸性对铁基离子液体脱硫过程的影响BmimFeCl4同时具备B-L 酸特性,目前普遍认为Bmim 环中 的C2-H 使其呈现出B 酸的特性,而L 酸特性则由[FeCl4]-产生。Wang 等[78]应用吡啶红外光谱来表征不同摩尔比M[n(FeCl3·6H2O):n(BmimCl)]合成的铁基离子液体的B-L 酸度变化规律,其中L 酸的酸度随M的增加而增加,当M降低至0.3 时,含有Fe(Ⅲ)的离子液体仅表现出Lewis酸性。
偏碱性的非质子有机溶剂(DMF、DMI)组成的复合脱硫体系对硫化氢的吸收能力明显优于中性非质子溶剂NHD 复合体系。碱性溶剂较容易通过化学吸收H2S 形成HS-,而H2S 分子在中性或酸性溶剂中通过氢键的吸收能力较弱,难以解离。铁基离子液体脱除硫化氢的过程实质上是一个补酸的过程,氢离子浓度的增加增强了脱硫液的电导率、离子扩散速率以及氢键作用[66]。最终,H+与[FeCl4]-中Cl-结合,消弱了铁基离子液体阴阳离子间的相互作用,在促进脱硫反应进行的同时,又生成了HCl 而逸出体系,造成设备腐蚀[78]。
2.1.5 温度对铁基离子液体脱硫过程的影响 铁基离子液体的热稳定性主要与阳离子和阴离子的结构相关,BmimFeCl4在473 K 下结构与物化性能稳定[50](图3)。有文献报道,铁基离子液体的阴离子[FeCl4]-在338 K 时会以FeCl3、[FeCl2]+与Cl-形式存在[79],如果有H+引入则产生HCl[78]。实际上,非质子溶剂如DMAC、DMI 和NHD 等不仅在工业上得到广泛应用而且具有高沸点和高热稳定性的特点,与铁基离子液体按比例混合的复合液既保留了原组分良好的热稳定性又对铁基离子液体的结构和物化性能起到保护和优化作用。基于铁基离子液体BmimFeCl4的Nasil 工艺就可以在高温180℃以下的条件下运行,完全满足130~150℃条件下的直接熔硫要求。虽然高温不利于气体的吸收,但是温度升高会显著降低铁基离子液体自身的黏度,反而有利于气液间的传质过程[80-82],并且对于H2S 与CO2混合体系,升高温度有助于H2S的选择性吸收[83-84]。
表2列举了温度对铁基离子液体脱硫性能的影响情况。在283~453 K 处理低浓度硫化氢(1%~5%)时升高温度更有利于硫化氢的吸收,马云倩等[36,52]基于有机胺型铁基离子液体,考察了温度对脱硫效率的影响,对于Et3NHFeCl4,升高温度时脱硫效果也随之上升,与BmimFeCl4的规律一致。然而处理高浓度H2S(99.9%),气液间的传质过程受限制,较高的温度对脱硫不利。升温导致了铁基离子液体阴阳离子间距增大,Fe-Cl 键长变短[79],抑制了Fe(Ⅲ)的还原,致使其吸收转化速率降低。
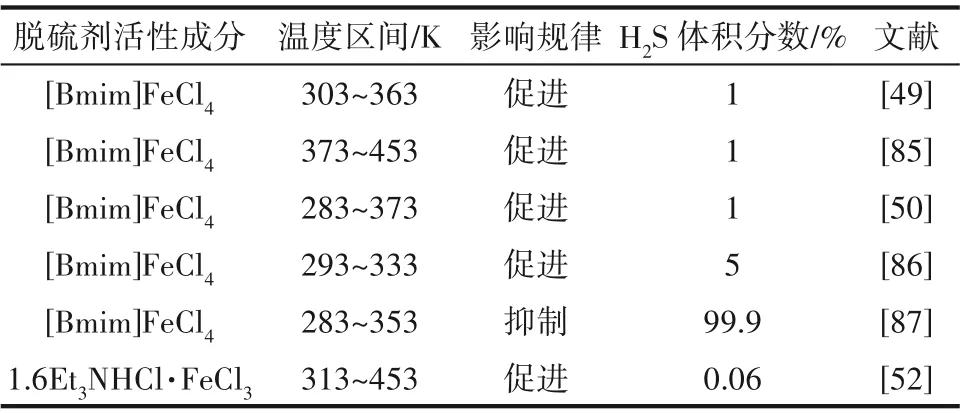
表2 温度对不同体系脱硫性能的影响Table 2 The effect of temperature on desulfurization performance in different systems
2.1.6 压力对铁基离子液体脱硫过程的影响 对于有气体参加的反应,增加压力能够提升反应速率,有利于H2S 的吸收,而压力对离子液体吸收H2S的影响,主要与离子液体的阴离子结构相关。此外,H2S在多数情况下与CO2共存,而压力能够显著影响离子液体对H2S/CO2的选择性吸收。Sakhaeinia 等[88]通过[Emim][PF6]和[Emim][Tf2N]中的溶解度和热力学函数进行分析,在一定压力下,咪唑基离子液体中的阴离子对H2S 溶解度的影响程度高于阳离子。Huang 等[84]指出咪唑基离子液体中H2S 的溶解度随压力的增大而增大,尤其在低压区,压力增大,溶解度迅速增大。当H2S 分压超过0.3 MPa,温度为333.15 K 时,H2S 在[Emim][Ace]中 的 溶 解 度 约 为2 mol/kg,远 高 于[Emim] [PF6] 中H2S 的 溶 解 度(0.11 mol/kg)[88]。Huang 等[83]和Wang 等[89]的 研 究 表明,低压和高温导致CO2容量的降低,有利于提高H2S+CO2混合体系中离子液体对H2S 的选择性。体系压力过高,虽能增加硫容,但受到反应动力学的制约,吸收溶解的硫化氢转化率却明显下降。研究表明,2.4 MPa 条件下H2S 在Fe-IL/NHD 中的平衡溶解度为2.86 mol/L,H2S 转化率仅为4.2%,降低压力到0.2 MPa时,H2S的转化率上升至13.2%[54]。
2.1.7 反应器类型对铁基离子液体脱硫过程的影响 开发高效反应器以强化气液传质是硫化氢脱除工艺强化的关键部分,特别是黏度较高的反应体系。目前对铁基离子液体脱硫实验室小试研究以鼓泡反应器为主[78],以探究脱硫反应机制和参数优化。Fe-IL复合体系脱硫的宏观动力学表现为快速拟一级反应[62],属于气相传质控制,处理低浓度含硫废气时使用喷淋吸收装置[90],能够很好地满足H2S的脱除。
动力波反应器(dynamic wave) 常用于气体吸收,见图6(a),待吸收气体自上而下进入逆喷管中与自下而上由喷头喷射进入的吸收液逆向接触,迫使液流沿径向呈辐射状自内向外射向器壁,最大限度地实现了高效传质的过程。李光晓等[91]定义了有效传质区(气液接触、传质过程中形成的两相流中气含率75%≤εg≤85%的区域),建立了有效传质体积的计算表达式。陈常蕊等[92]指出逆喷时的脱硫效率明显高于顺喷,说明动力波反应器有强化吸收的特性。葛喜乐等[93]采用动力波反应器作为铁基离子液体的脱硫塔,操作液气比为17 L/m3时动力波反应器能够有效强化脱硫过程,脱硫效率达86.8%。
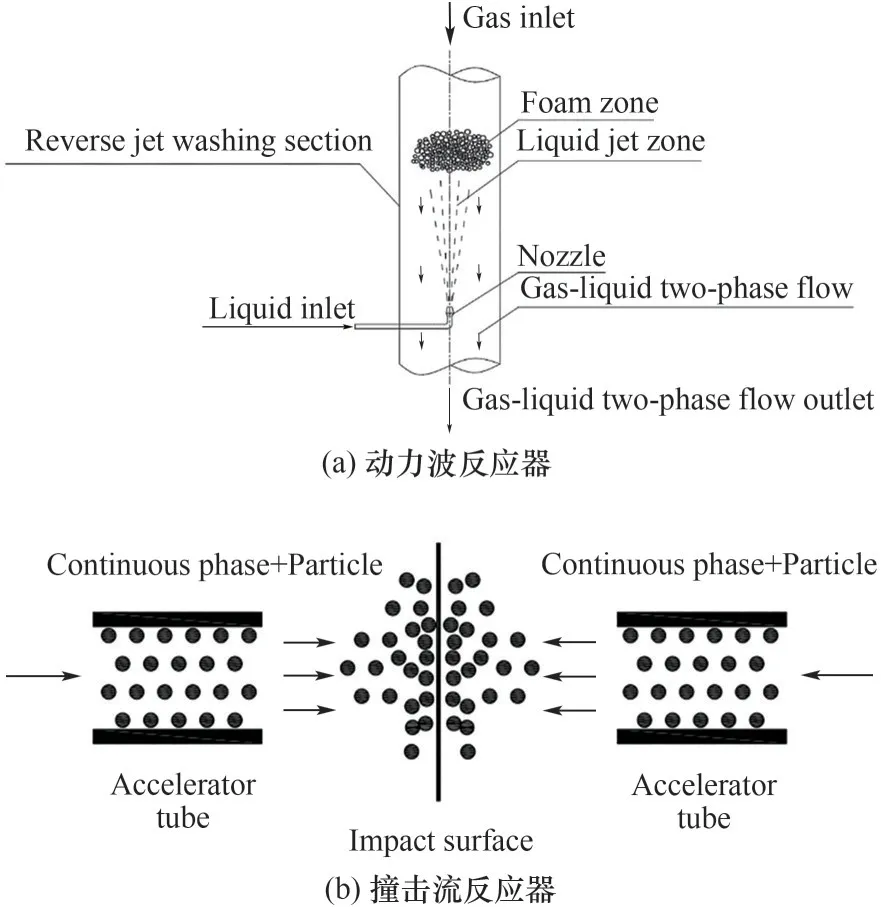
图6 气液混合传质强化方式Fig.6 Enhanced ways of gas-liquid mixing mass transfer
撞击流(impinging stream)作为一种传质过程强化技术,可以使气液相间有更好的微观混合,原理见图6(b),两股气液混合流体进行反向同轴高速碰撞,强化传热传质过程,目前撞击流反应器在气-液传质领域都有广泛的应用[94]。Wu 等[95]以Ca(OH)2悬浮液为脱硫剂通过撞击流吸收烟道气的SO2,并计算其传质系数;李发永等[96]基于撞击流反应器,应用去离子水进行CO2的吸收,对撞击流吸收器、喷射式吸收器和鼓泡式吸收器进行对比,发现液气比相同时,撞击流吸收器的传质系数和CO2吸收率要高于另外两种反应器。2015 年,唐山三友集团将气液撞击流反应器应用于60000 m3/h 含H2S 工业废气的脱除,将H2S 浓 度 由2000 mg/m3降 低 至10 mg/m3以下[94]。然而,目前基于撞击流反应器强化铁基离子液体湿式氧化脱硫工艺的研究暂未见报道。
2.2 脱硫再生反应
2.2.1 氧化还原介体对铁基离子液体再生过程的影响 氧化还原介体(ROMs)可以加速氧化还原反应过程中的电子传递速度,能够有效提升其反应速率[97-99]。氧化还原介体可在亚铁离子氧化过程中,优先与亚铁离子反应,被还原后的氧化还原介体可以较为容易地通过氧气实现氧化再生。此过程可在不活化氧的条件下提高再生效率,避免脱硫体系中活化氧物种的过度氧化反应发生。目前常见的氧化还原介体包括蒽醌-2,6-二磺酸钠(AQDS)、蒽醌-2-磺酸钠(AQS)[99]、腐殖酸以及多种价态过渡金属盐[Cu(Ⅰ/Ⅱ)][68]等。在硫化氢脱除领域,ADA法中的催化剂便采用了蒽醌-2-磺酸钠,借此活化体系中金属V 对硫化氢的氧化还原能力[13],余江等[68]向铁基离子液体脱硫富液中引入铜基离子液体[Cu(Ⅱ)-IL],利用铜基离子的中间价态来强化铁基离子液体的氧化还原性能,显著地提升了脱硫液中的脱硫再生效率,其反应机理见图5。
2.2.2 电解法对铁基离子液体再生过程的影响 可以通过无隔膜电解法实现铁基离子液体脱硫富液中的Fe3+/Fe2+和H+/H2离子对的转化而达到再生脱硫液的目的。但由于H+浓度很低,Guo 等[58]采用6 mol/L HCl 溶液作为氢池,使用隔膜电解法对铁基离子液体脱硫富液进行电化学反应再生,在亚铁离子氧化为三价铁离子的同时,将脱硫富液中的氢离子还原成氢气,使硫化氢中的硫元素和氢元素得到了全资源化利用。但是电解过程中由于脱硫液中Cl-浓度较高,需要提升析氯电位,降低析氯反应对Fe(Ⅱ)氧化的干扰。如何富集提高脱硫富液中的H+浓度,以及开发无氯铁基离子液体脱硫剂,需要从脱硫剂的结构设计及脱硫工艺方面进一步探索[100-101]。非水相湿法氧化脱硫电解再生联合产氢这一工艺符合新能源及清洁生产的需求,亟需深入研究。
2.2.3 曝气方式对铁基离子液体再生过程的影响射流曝气和采用气体分布器是湿法氧化脱硫再生工艺中常用的曝气技术。微气泡(直径为1~100 μm)强化氧化技术近年来被广泛应用到环境领域。研究表明,水相中表面带大量负电荷的微气泡会自发地收缩并最终爆裂,在局部的高温高压的作用下,气泡界面上富集的高浓度离子发生反应,瞬间释放出积蓄的大量化学能,激发产生大量活性氧负离子和自由基,显著提高介质氧化能力[102-103]。王亚茹等[104]应用微气泡技术来强化亚铁离子的氧化,取得了显著的效果,并研究了曝气头尺寸、反应温度、酸浓度等因素对亚铁离子的氧化效率以及羟基自由基生成的影响。微气泡由于比表面积大,气含率高,在非水相体系中十分有利于气液混合,促进气液两相反应的进行,避免可能的空化现象发生。
2.3 硫磺产物的分离与纯化
非水相金属基离子液体脱硫工艺中脱硫富液与脱硫产物硫磺可以采用过滤、离心或沉降的方式分离硫磺并回收离子液体。分离所得的硫膏或硫浆熔硫处理得到液体硫磺与脱硫液两相体系,底部分离出的熔融态硫磺经冷却结晶后得到固体硫磺产物,上部分离的脱硫液可循环使用,避免了水相湿法脱硫体系难熔硫和脱硫液降解产生废水的局限性(图7)。一般情况下,小试熔硫温度控制在115~125℃,产物硫磺纯度≥99.0%[56],现场实验所得硫浆或硫膏可以在130~150℃完成熔硫。研究表明,向脱硫体系中引入有机溶剂可降低硫磺在铁基离子液体中的溶解度,其中添加33%的DMI 溶剂对硫磺的析出十分有利。此外,复配有机溶剂能够进一步地降低体系的黏度与密度,加快硫磺沉降的速率,利于熔硫后的分相[54,56]。表面活性剂的引入同样可以降低硫磺的溶解度,并且还能够明显促进小颗粒的团聚[105]。杨溢等[106]采用1%的TX-100(非离子型表面活性剂)诱导硫磺颗粒进行团聚,在其作用下,小颗粒间凝聚形成水化层,阻止硫磺颗粒进一步溶解于铁基离子液体中。加入TX-100 后,产物硫磺的平均粒径由1.101 mm升高至3.041 mm。
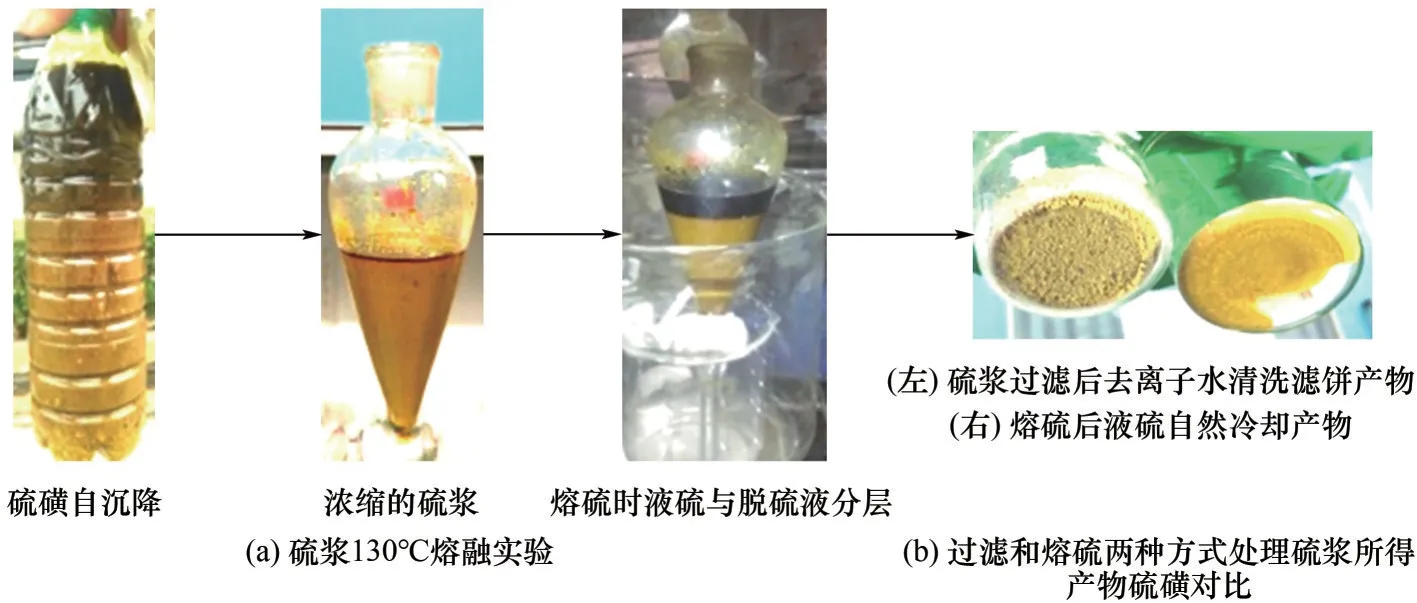
图7 熔硫过程实验现象以及硫磺产品形貌Fig.7 The experimental phenomena of sulfur melting process and morphology of sulfur products
2.4 原料气杂质组分影响
原料气涉及行业众多,其中最为关注的是CO2和NH3的影响。天然气及生物沼气中存在着大量的二氧化碳,而以Lo-Cat 为代表的传统水相湿式氧化脱硫技术,由于是碱性体系,对酸性气体选择性差,难以调控,并且极易产生大量高盐废水[22]。而Nasil工艺由于其是酸性体系,能够氧化硫化氢,而无法与二氧化碳反应,因此选择性较强;此外,Fe-IL 对二氧化碳有一定的物理吸收能力,25℃、1.2 MPa下,吸收液中CO2浓度可达0.6436 mol/L,复配有机溶剂DMAC 后,浓度上升至0.8195 mol/L[53]。张晓东[53]等开发了一种绿色脱硫脱碳工艺,以铁基离子液体复配有机溶剂作为吸收剂,首先通过与原料气接触,实现硫化氢的转化,随后脱硫富液经过闪蒸解吸出溶解态的CO2,该工艺有利于实现我国碳达峰、碳中和的目标。
对于焦炉煤气中NH3的影响往往无法忽视,水相HPF 碱性脱硫体系中,氨气逸出的现象十分严重[14]。而BmimFeCl4兼具Lewis 酸和Brönsted 酸的特性,使其既具备氧化能力,又能够与碱性气体发生中和反应[50]。因此,余江等[107]提出了一种利用BmimFeCl4同步脱硫脱氨的工艺,利用NH3与[Bmim]+中的C2-H 发生的酸碱中和反应,生成了NH4Cl 沉淀,实现了脱氨的目的,氨容达到26.35 g NH3/L 以上,脱氨效率达99.99%。脱氨过程中消耗的HCl 可以使用HCl 气体、盐酸溶液或FeCl3·6H2O为原料再生金属基离子液体,然后循环使用。
BmimFeCl4对小分子碳氢化物如CH4吸收能力很弱,即使与有机溶剂复配之后如BmimFeCl4/DMAC复合体系,增压对CH4的吸收没有明显效果[53]。He 等[108]应用BmimFeCl4对小分子烃类物质以及二氧化碳进行吸收,发现气体溶解度由大到小依次为环丙烷>丙烯>丙烷≈二氧化碳,指出BmimFeCl4更有利于吸收环状气体分子。
2.5 新型非水相脱硫工艺的构建
基于铁基离子液体的非水相脱硫工艺正逐步完善,推向工业应用,该工艺流程通式如图8 所示,原料气首先经过三相分离器进行预处理,除去水分、重烃类物质以及固体杂质,随后进入到吸收塔中,与来自再生塔的脱硫液逆流接触,实现硫化氢的高效脱除,此时硫化氢被氧化为硫单质,净化后的原料气进入后续工段。吸收硫化氢后的脱硫富液经过富液泵进入闪蒸罐,分离出CO2和少量溶解在脱硫剂中的烃类物质,闪蒸后的脱硫富液进入再生塔,进行脱硫液的再生。再生方式可以是空气曝气和电解再生两种方式,再生后的脱硫液夹带着硫磺进入沉降槽中进行硫磺的沉降,沉降罐底部的固液混合物经熔硫釜熔硫后,得到产品硫磺,而熔硫釜分离出的液体与沉降罐上清液共同汇至再生塔中。

图8 金属基离子液体脱硫工艺流程图Fig.8 The flow chart of desulfurization process with metal-based ionic liquid
工业示范应用表明[12],非水相金属基离子液体脱硫工艺操控简单,脱硫过程中无须添加化学辅剂和消耗水资源,运行能耗低,硫磺产品纯度高、无二次污染,符合小型撬装工艺和设备的要求,适合偏、散和储量变化大的气田开采以及生产工艺段的废气处理。如果采用电解再生,可以生产氢清洁能源。
3 金属基离子液体湿法氧化脱硫工艺面临的挑战
3.1 脱硫液组分配伍
何义等[49]报道的BmimFeCl4在常压室温条件下的硫容是0.31 g/L,侧线实现验证结果表明,由BmimFeCl4与有机溶剂复配组合的复合离子液体脱硫液的硫容仅为1 g/L,与脱硫液中实际铁含量所能氧化硫化氢的计量反应数值相去甚远。从分子结构上分析,铁基离子液体中阴离子[FeCl4]-属正四面体结构[67],空间位阻的作用不利于三价铁离子与硫化氢分子之间发生氧化还原反应。因此如何有效地活化脱硫体系中的催化氧化中心,成为了现阶段Nasil 工艺的关键科学问题。王建宏等[46,109]报道了使用络合铁基离子液体的水相脱硫工艺,[FeEDTA]-的活性中心更容易与硫化氢发生轴向相互作用,表现出较好的脱硫性能。此外,螯合金属基型离子液体在处理含碱性气体组分的原料气时,与H2S 分子间的氢键与范德华力起到强化促进吸收作用,且螯合金属离子具有的化学稳定性有助于避免脱氨过程中脱硫液损耗与二次污染等问题。
3.2 脱硫过程酸平衡控制
复合铁基离子液体脱硫液的酸性主要来自两个方面,一个是阳离子咪唑环上的C2-H,其次是在脱硫阶段硫化氢分子被氧化为硫磺后释放的H+。已经报道铁基离子液体的阴离子[FeCl4]-实际以多种复合离子形态存在,受温度影响明显,其中游离态Cl-与脱硫液中的H+结合可形成HCl,容易在脱硫过程富集,然后随气流以HCl 气逸出[78]。如果体系中含有一定水分,游离态的氯离子容易腐蚀金属设备。非质子溶剂如N-甲基二乙醇胺(MDEA)、N,N-二甲基乙酰胺(DMAC)等与铁基离子液体BmimFeCl4复配后可起到束缚HCl作用,且能促进硫磺产物颗粒的长大[53]。为了从根本上解决铁基离子液体非水相脱硫过程中的酸流失和减轻设备腐蚀,开发无氯离子的络合型金属基离子液体脱硫剂很有必要。
3.3 再生过程强化
脱硫富液的再生效率直接影响了非水相铁基离子液体脱硫工艺Nasil 运行的长效性以及产物硫磺的品质。实验表明,铁基离子液体具备良好的吸氧储氧能力,然而脱硫富液中Fe2+氧化为Fe3+再生效率较低。研究发现,溶于水相后的铁基离子液体能活化空气中的氧气,表现出强的氧化能力[57],实际上这与Lo-Cat 体系的氧气因与铁离子发生芬顿反应而被活化的机制是一致的。由此可见,非水相铁基离子液体中的O2分子与铁离子之间的作用方式有很大差别。基于高沸点有机溶剂与金属基离子液体复合体系组成的非水相湿法氧化脱硫工艺,提升溶解氧分子对亚铁离子的氧化效率成为Nasil 工艺提高脱硫效率的关键,需要深入研究铁基离子液体中氧化中心铁离子结构-非质子溶剂-O2分子之间的相互作用及调控机制。
3.4 其他类型非水相脱硫体系
Liu 等[110-111]尝试将铜纳米颗粒与低共熔溶剂(DES)结合制备出了非水相纳米流体来进行硫化氢的脱除,结果表明其具备优越的脱硫性能,但该体系的循环性较差,四次循环后该溶剂的稳定性和循环性明显降低。Wang 等[112]发现杂多酸离子液体[Bmim]PMo10V2O40/BmimCl在200℃下,能够展现出较好的脱硫性能,并且能够通过鼓入空气进行再生;Ma等[113]的研究表明BmimCl中的Cl-与H2S存在较强的相互作用,因此Cl-进入到PMo12O3-40的空腔后,能够显著增强其脱硫性能。
4 展 望
本文总结了铁基离子液体湿式氧化脱硫技术十余年的研究进展,评述了铁基离子液体脱硫性能及Nasil 工艺的特点,对此技术的发展提出如下建议。
(1)非水相金属基离子液体湿法氧化脱硫概念的深入与发展
将金属基离子液体与非质子溶剂复合所组成的非水相脱硫体系,集酸性、氧化还原可逆性、高热稳定性于一体,已经展现出良好的氧化脱硫性能和实用性,能针对性地解决水相湿法氧化脱硫所存在的严重二次污染的瓶颈问题。目前非水相金属基离子液体脱硫体系的研究主要局限于宏观层面对脱硫体系的优化,离子液体中离子对的形成与形态研究,以及离子对活化金属基离子液体催化性能的反应机制的相关报道较少。理论计算与实验研究相结合,研究氧化脱硫过程中阴阳离子的迁移规律与结构重组的本征规律,建立非水相氧化脱硫体系中的脱硫反应与再生强化调控机制,为非水相金属基离子液体湿法氧化脱硫概念的发展以及新型络合型金属基离子液体的研发提供分子层面的理论支撑。
(2)非水相金属基离子液体湿法氧化脱硫工艺的引领作用
利用金属基离子液体可溶解性和可设计性的优点,容易调控脱硫体系中活性成分的含量和结构,提升脱硫液的氧化脱硫能力;高沸点非质子溶剂可消除空化效应及氧气活化现象,避免产生气蚀和活性氧的过度氧化副作用;活性成分及高沸点溶剂的高热稳定性特性有利于适应宽泛的脱硫反应温度,可以实现直接熔硫,获得高纯度硫磺,表现出独特的绿色脱硫优势。金属基离子液体氧化脱硫工艺的绿色高效符合分散式撬装化脱硫工艺的要求,是一种极具潜力的新型脱硫技术。从该工艺的关键技术问题出发,优化脱硫体系,完善脱硫工艺,有利于工业化应用的拓展。
(3)非水相湿法氧化脱硫技术对清洁能源发展的助推作用
铁基离子液体脱硫富液再生过程直接决定了该工艺的可循环性与产品硫磺的品质,电解法可使硫化氢中的氢元素实现资源化,满足清洁生产的要求。Nasil 工艺的非水介质特性避免了电解过程中H2O 的干扰,通过富集脱硫富液中H+和Fe2+浓度提高电解效率,可实现脱硫富液的高效再生,并对清洁氢能的发展起到助推的作用。
猜你喜欢
化工管理(2022年31期)2022-12-23
中国化肥信息(2022年8期)2022-11-30
中国化肥信息(2022年5期)2022-08-30
中国化肥信息(2022年4期)2022-06-07
中国化肥信息(2022年2期)2022-04-19
农业研究与应用(2021年3期)2021-08-23
能源工程(2021年1期)2021-04-13
矿业安全与环保(2021年1期)2021-03-06
燃料化学学报(2020年3期)2020-05-07
劳动保护(2018年11期)2018-10-26
