
肿瘤相关成纤维细胞在胰腺癌治疗耐药中的作用
2022-05-14 01:25贾军梅郭亚荣
临床肝胆病杂志 2022年5期
闫 毅, 贾军梅, 郭亚荣
1 山西医科大学 第一临床医学院, 太原 030001; 2 山西医科大学第一医院 肿瘤科, 太原 030001
胰腺癌最常见的病理类型是胰腺导管腺癌(pancreatic ductal adenocarcinoma, PDAC),恶性度高,5年生存率仅8%[1]。以吉西他滨为主的化疗方案,仍是大多数胰腺癌患者的主要治疗方法。然而,化疗给患者带来的疗效有限,中位无进展生存期仅为6.8个月[2];免疫治疗在其他恶性肿瘤的治疗上取得了实质性的成功[3],然而在PDAC中的临床获益却非常有限[4]。这都归因于胰腺癌致密的间质,胰腺癌间质占胰腺肿瘤体积的90%,包括成纤维细胞、免疫细胞、炎症细胞、生长因子、细胞外基质(ECM)等[5]。其中,肿瘤相关成纤维细胞(cancer-associatedfibroblasts, CAF),即活化的胰星状细胞(PSC),是PDAC间质的主要细胞成分,其可产生过量的ECM成分[6]。越来越多的证据[7-8]表明CAF是胰腺癌化疗、放疗、靶向治疗、免疫治疗耐药的主要参与者,尤其是在化疗和免疫治疗中。本文主要从化疗和免疫治疗两个方面论述CAF在胰腺癌治疗耐药中的作用机制。
1 化疗耐药
化疗是胰腺癌最重要的治疗手段之一,CAF限制抗肿瘤药物的输送,影响抗肿瘤药物的代谢,并调节癌细胞的凋亡,在化疗耐药中起重要作用(图1)。

图1 CAF参与化疗耐药
1.1 阻碍化疗药物的输送 化疗药物需要穿过ECM才能进入癌细胞的细胞质,发挥抗癌作用。胰腺癌细胞激活CAF,使得CAF增殖并产生ECM蛋白,在癌细胞周围形成阻止化疗药物进入的屏障[9]。与此同时,激活的CAF产生高分子量透明质酸[10],透明质酸过度表达并在间质中沉积,通过形成凝胶-流体相产生间质高压力,导致广泛的血管塌陷和低灌注,进一步阻碍了化疗药物的输送[11]。靶向抑制CAF分泌的βig-h3蛋白,可以通过改变M2型巨噬细胞分泌的炎症和细胞毒性分子,重新编程肿瘤微环境,降低间质压力[12]。除了在基质结构和间质压力中的作用,Hessmann等[13]发现了CAF诱导的药物清除现象,即:CAF将活性吉西他滨包裹在细胞内,导致药物清除,从而隔离化疗药物,限制肿瘤细胞获取吉西他滨。 这一发现揭示了CAF在PDAC中形成间质屏障的新机制。
1.2 干扰抗癌药物的摄取及细胞内代谢 吉西他滨是一种脱氧胞苷核苷类似物(2′,2′-difluorodeoxycytidine, dFdC),是目前PDAC化疗的一线治疗药物[14],通过细胞膜上表达的平衡核苷转运蛋白1(HENT1)和浓缩核苷转运蛋白3(HCNT3)摄取后进入癌细胞的胞浆,被胞内的磷酸化限速酶脱氧胞苷激酶连续磷酸化,最终生成吉西他滨的活性代谢物dFdC三磷酸,抑制DNA复制发挥细胞毒作用[15]。
CAF通过影响吉西他滨摄取和代谢关键酶的表达,降低吉西他滨的细胞毒性。第一,与人PDAC来源的原代癌细胞和PDAC细胞系相比,CAF对吉西他滨的细胞毒作用具有固有的抵抗力,这是因为CAF中hENT1和DCK的表达显著降低,使得CAF对吉西他滨的摄取及其代谢能力降低[16];激活的CAF过度表达TGFβ,通过TGFβ-ALK5-Smad2/3 信号通路诱导CAF中富含半胱氨酸蛋白61(CYR61)的表达,而CYR61是两种重要的核苷转运蛋白hENT1和hCNT3的负性调节因子,从而削弱肿瘤细胞对吉西他滨的摄取[17]。第二,CAF通过平衡核苷转运蛋白(ENT)分泌大量脱氧胞苷,脱氧胞苷不会阻碍吉西他滨进入细胞,而是在肿瘤细胞内竞争关键酶脱氧胞苷激酶的磷酸化作用,抑制PDAC细胞对吉西他滨等核苷类似物的代谢,因此降低了吉西他滨的抗肿瘤作用。值得一提的是,并不是所有CAF都具有分泌脱氧胞苷的特性,而是其中一种表型具有的特性,这显示了CAF的异质性[18]。
1.3 调控癌细胞凋亡相关信号
CAF影响PDAC细胞化疗耐药的内在机制,CAF激活肿瘤细胞抗凋亡的相关信号通路,降低吉西他滨诱导的细胞凋亡水平,使肿瘤细胞能够在细胞毒性药物的影响下存活[19]。
1.3.1 CAF分泌ECM蛋白激活抗凋亡信号通路 CAF分泌纤维连接蛋白(FN)促进激活ERK1/2,从而保护肿瘤细胞免受化疗诱导的凋亡[20]。CAF表达钙黏蛋白11,对胰腺癌肿瘤细胞耐药发挥一定的作用;敲除或抑制钙黏蛋白11可以减少胰腺肿瘤的生长,增加它们对吉西他滨的反应[21]。PDAC细胞分泌的谷氨酰胺转氨酶(TG2)激活CAF并分泌层粘连蛋白A1,层粘连蛋白A1诱导局灶性黏附激酶(FAK)的磷酸化,激活肿瘤细胞内PI3K-Akt途径保护癌细胞免受吉西他滨等化疗药物的细胞毒性作用[22]。
1.3.2 CAF分泌多种生长因子和促炎成分激活抗凋亡信号通路 CAF分泌SDF-1(CXCL12)与胰腺癌细胞中的CXCR4特异性结合,上调胰腺癌细胞中SATB-1(一种位于染色体3p23上的核基质结合区结合蛋白)的表达,并诱导IL-6自分泌环路,诱导胰腺癌细胞对吉西他滨产生耐药性,并维持CAF的表型,导致纤维炎性肿瘤微环境的恶性特征[23-24]。CAF产生的肝细胞生长因子(HGF)通过旁分泌可以激活癌细胞中的c-Met/PI3K/Akt通路,从而抑制癌细胞凋亡并诱导对吉西他滨的化疗耐药[16]。进一步研究[25]发现,CAF中Prrx1的表达可以调控HGF的分泌,Prrx1低表达的CAF分泌HGF减少,肿瘤细胞对吉西他滨的治疗更敏感。CAF通过SMAD2/3途径分泌TGFβ1来上调PDAC细胞中ATF4的表达,ATF4直接与ABCC1的特异性启动子区域结合并激活其转录,最终导致吉西他滨耐药[26]。CAF来源的胰岛素样生长因子(IGF)通过激活癌细胞内的胰岛素/IGF1R通路来钝化化疗反应[27]。KRAS突变的胰腺肿瘤细胞及CAF分泌IL-1β,激活IL-1β-IRAK4环路,驱动肿瘤细胞内的NF-κB活性,促使间质纤维化及肿瘤细胞对化疗耐药[28]。
1.3.3 CAF与肿瘤细胞相互作用 胰腺癌细胞的主要驱动基因,如KRAS、CDKN2A、TP53和Smad4的特定突变,也可以影响CAF的分泌体和表型,进一步促进化疗耐药。如:与p53缺失细胞相比,突变p53的胰腺癌细胞存在时,CAF表现出更强的侵袭、转移和耐药特性。从机制上讲,NF-κB信号在p53突变的胰腺癌细胞中增强,从而刺激CAF中HSPG2(Perlecan)的旁分泌,由此导致了CAF恶性表型并延迟了体外和体内的化疗反应。此外,在含有癌细胞和CAF异质性亚群的胰腺肿瘤中,fl-e-CAF(分离自p53缺失型肿瘤的CAF)可以通过与mt-CC(p53突变型癌细胞)或mt-e-CAF(分离自p53突变型肿瘤的CAF)相互作用而被重新编程,从而使更多的CAF获得恶性表型,造成肿瘤的侵袭、转移和耐药[29]。
1.4 化疗诱导的促肿瘤反应 化疗能够诱导促肿瘤反应,是肿瘤获得性耐药的主要机制,为治疗有效后出现进展提供了合理的解释。吉西他滨治疗会引起CAF的分子变化,化疗后的CAF(R-CAF)比未治疗的CAF(N-CAF)更支持肿瘤细胞的存活、迁移和侵袭。这是由于R-CAF驱动应激相关MAPK信号的促炎反应,诱导多种炎性介质SASP(衰老相关分泌表型)表达上调,从而增强肿瘤细胞的生长和侵袭性[30]。此外,化疗诱导CAF分泌外泌体miRNA-106b,进一步增强了CAF对吉西他滨的耐药,同时吉西他滨能够上调CAF中miR-106的表达,miR-106转移到胰腺癌细胞,导致肿瘤抑制基因TP53INP1的表达减少和癌细胞的增殖[31]。
1.5 CAF调控肿瘤干细胞促进化疗耐药 肿瘤干细胞被认为是造成胰腺癌治疗耐药的重要原因。CAF为肿瘤干细胞提供生存生态位,维持肿瘤干性,促进其化疗耐药潜能。CAF旁分泌细胞因子为胰腺癌干细胞构建生存生态位:CAF分泌的GPC4,激活Wnt/β-catenin通路,诱导肿瘤干细胞标志物(c-Myc、SOX2、MDR1)的表达,维持胰腺癌干细胞特性,造成胰腺癌5-FU治疗耐药。抑制GPC4增强了胰腺癌细胞对5-FU的敏感性,并抑制了干细胞样特性[32]。CAF分泌CXCL12也介导胰腺癌细胞与干细胞相互作用,E26转化特异性同源因子通过抑制CXCL12受体CXCR4的转录、降低胰腺癌细胞对肿瘤干细胞生态位刺激的敏感性,抑制肿瘤干细胞干性,使得PDAC对吉西他滨治疗敏感[33];CAF促进肿瘤干细胞的表型和功能:激活后的CAF,胶原合成增加,激活胰腺癌干细胞中的整合素β1-FAK信号转导,增加胰腺癌干细胞中乙醛脱氢酶的表达,促进胰腺癌干细胞表型,肿瘤细胞表现出更强的克隆生长、自我更新、迁移、耐药能力[34];CAF促进化疗诱导的肿瘤干细胞富集:随着化疗诱导的表型和功能改变,如CAF中持续的STAT-1和NF-κB活性,使得胰腺CAF分泌ELR+CXC类趋化因子,它们通过CXCR2与癌细胞结合,促使肿瘤细胞转分化为肿瘤干细胞表型,并促进治疗后的侵袭性行为[35]。
2 免疫治疗耐药
免疫治疗在PDAC中的临床获益非常有限。CAF形成物理屏障干预免疫治疗药物发挥作用,分泌免疫抑制因子、募集免疫抑制细胞的浸润、抑制免疫细胞的活化和功能,造成胰腺癌免疫治疗耐药(图2)。
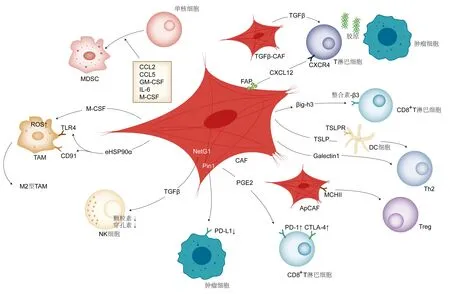
图2 CAF参与免疫治疗耐药
2.1 CAF干预非特异性免疫
2.1.1 CAF参与髓源性抑制细胞(MDSC)的募集和分化 Kuen等[36]在胰腺癌细胞、CAF和单核细胞的三维共培养模型的上清液中,检测到了高水平的粒细胞巨噬细胞集落刺激因子(GM-CSF)、巨噬细胞集落刺激因子(M-CSF)、IL-6、IL-10及CCL-2,其中GM-CSF、M-CSF、IL-6参与髓系细胞向MDSC分化和募集,IL-10诱导巨噬细胞M2型极化并抑制T淋巴细胞的功能。此外,胰腺肿瘤细胞分泌促炎细胞因子IL-1β,促进CAF的激活和分泌表型的形成,进而诱导炎症程序,上调细胞因子和趋化因子CCL2、CCL5,进而诱导MDSC、肿瘤相关巨噬细胞(TAM)的浸润[37]。MDSC的募集和分化可以抑制CD8+T淋巴细胞的浸润和功能,加剧免疫抑制微环境。靶向间质中的透明质酸,可以抑制CAF中CXCR4介导的信号轴,调节髓系细胞的功能,使PDAC中CD8+T淋巴细胞浸润的数量增加[38]。
2.1.2 CAF募集TAM并诱导促肿瘤表型 内皮-间质转化来源的CAF可以直接促进TAM的浸润,同时分泌较高水平的胞外热休克蛋白90α(eHSP90α),eHSP90α与巨噬细胞上的TLR4受体和CD91受体结合,激活其下游的MyD88JAK2/TYK2STAT-3途径,抑制巨噬细胞M1型极化,诱导巨噬细胞M2型极化,并激活巨噬细胞中HSP90α的前馈环路,使得巨噬细胞大量分泌和表达HSP90α,进一步诱导了TAM的浸润和M2型极化[39]。此外,CAF分泌M-CSF并增加单核细胞内活性氧的产生,进而诱导巨噬细胞M2型极化[40]。M2型巨噬细胞是低效的抗原提呈细胞,在肿瘤微环境中抑制T淋巴细胞的迁移、激活和增殖,发挥免疫抑制作用[41]。
2.1.3 自然杀伤(NK)细胞 NK细胞可以不依赖于抗原呈递而破坏PDAC细胞,是胰腺癌中极具治疗潜力的抗肿瘤免疫细胞类型[42]。CAF中高表达NetG1,通过与肿瘤细胞中的NGL-1结合,激活由AKT/4E-BP1、p38/FRA1、囊泡谷氨酸转运蛋白1及谷氨酰胺合成酶组成的信号通路。一方面,为肿瘤细胞提供关键代谢物,如Glu/Gln,帮助PDAC细胞在营养匮乏条件下存活;另一方面,分泌大量免疫抑制因子,如:GM-CSF、 IL-1β、 IL-8、 CCL20/MIP3α 及 TGFβ,尤其是TGFβ显著抑制NK细胞的激活和功能,并保护PDAC细胞免受NK细胞介导的死亡,形成了PDAC免疫抑制微环境。敲除CAF中的NetG1基因可部分通过IL-15消除其免疫抑制表型,使得NK细胞发挥抗肿瘤功能[43]。此外,TGFβ也可以通过减少NK细胞的激活受体、颗粒酶B以及穿孔素,抑制NK细胞的激活和细胞毒活性[44]。
2.2 CAF干预特异性免疫
2.2.1 CAF抑制T淋巴细胞瘤内浸润 CAF产生FAK增加肿瘤环境中的胶原沉积,形成T淋巴细胞浸润肿瘤的物理屏障[32]。CAF中的NF-kB介导的CXCL12分泌通过阻止细胞毒性T淋巴细胞渗透到肿瘤中,促进肿瘤免疫逃逸和肿瘤生长[45]。Dominguez等[46]确定了一组由转化生长因子β驱动的CAF亚群(TGFβ-CAF),并确定是晚期胰腺肿瘤中的主要成纤维细胞类型,TGFβ-CAF的高表达与免疫抑制剂治疗反应差相关。抑制TGFβ可以降低CAF活性,进而减少间质纤维化并增加T淋巴细胞浸润,最终改善免疫治疗效果[47]。
2.2.2 CAF减少T淋巴细胞的增殖和活化 CAF分泌βig-h3,βig-h3通过与CD8+T淋巴细胞上高表达的整合素β3结合,导致Hic-5蛋白与Y505磷酸化的淋巴细胞特异性蛋白酪氨酸激酶结合增加,钝化信号转导,减少肿瘤特异性CD8+T淋巴细胞的增殖和活化,使胰腺肿瘤细胞逃避免疫清除。在KIC小鼠体内,抑制βig-h3蛋白的表达,与抗PD-1抗体联合治疗具有协同作用[12]。
2.2.3 CAF刺激CD4+T淋巴细胞分化为更多的免疫抑制亚型:调节性T淋巴细胞(Treg细胞)和Th2细胞 Elyada等[48]发现了新的CAF亚群:抗原呈递CAF(apCAF),它表达MHC II类分子,并能够在体外向CD4+T淋巴细胞呈递模式抗原。然而,apCAF缺乏诱导T淋巴细胞增殖所需的共刺激分子,使得apCAF表达的MHCⅡ类分子作为受体,诱导CD4+T淋巴细胞分化为Treg,降低CD4+T淋巴细胞抗肿瘤免疫能力。相比正常胰腺成纤维细胞,胰腺癌成纤维细胞表达更高水平的Galectin 1,且在共培养中诱导更高水平的T淋巴细胞凋亡,此外,胰腺癌成纤维细胞激活线粒体凋亡途径中的半胱氨酸天冬氨酸蛋白酶9及半胱氨酸天冬氨酸蛋白酶3,并刺激Th2型细胞因子(IL-6和IL10)的分泌,减少Th1型细胞因子(TNFβ和IFNγ)的分泌,导致免疫偏移[49]。Gal1基因缺失会上调肿瘤细胞中浸润的CD4+和CD8+T淋巴细胞[50]。胸腺基质淋巴生成素(TSLP)是CAF在肿瘤源性IL-1α、IL-1β及 ASC的作用下分泌的,活化的CAF同时上调树突状细胞(DC)上的TSLP受体(TSLPR),分泌Th2细胞趋化因子,诱导TSLP依赖的Th2极化[51]。
2.2.4 CAF作用于免疫检查点及其配体 CAF上调T淋巴细胞上免疫检查点,CAF通过PGE2促进T淋巴细胞上免疫检查点TIM-3、PD-1、CTLA-4和LAG-3的表达,降低了CD8+T淋巴细胞HLA-DR的表达,另外表达免疫检查点的增殖T淋巴细胞在重新刺激后产生的IFNγ、TNFα和CD107a较少,这意味着T淋巴细胞效应器功能的丧失。同时,PD-1的上调会进一步抑制CD8+T淋巴细胞的增殖[52];CAF降低PD-1配体的表达,Pin1在CAF中过表达,驱动促结缔组织增生,作用于HIP1R中的pSer929-Pro基序促进癌细胞中PD-L1的内吞和溶酶体降解,造成PD-L1表达降低,使得PDAC对免疫治疗低应答[53]。
2.3 成纤维细胞活化蛋白(FAP)的表达干预免疫治疗耐药 FAP是选择性表达于CAF的一类膜糖蛋白,作为激活CAF的重要标志物之一。肿瘤细胞分泌的TGFβ可以诱导CAF表达FAP,高水平的FAP与胰腺癌患者较差的预后相关[54]。FAP参与胰腺癌细胞逃避免疫监视,FAP高表达的CAF是趋化因子CXCL12的主要来源[55]。此外,FAP抑制免疫细胞功能,耗尽FAP可以降低免疫抑制性细胞(MDSC、TAM)产生精氨酸酶-1(Arg1)和诱导型一氧化氮合酶的能力,进而阻碍MDSC和TAM对CD8+T淋巴细胞功能的抑制,同时通过降低肿瘤微环境中的细胞因子/趋化因子,如IL-10、TGFβ、CCL5和CCL22,抑制免疫抑制性细胞(ISC)内STAT6信号通路的活性,减少肿瘤浸润ISC的数量和免疫抑制功能[56]。
3 结语
综上所述,CAF作为胰腺癌间质的主要成分,在胰腺癌治疗耐药中发挥重要作用。随着生物技术的发展,更多新的CAF亚型被定义,例如:近期Wang等[57]在间质疏松(低结缔组织增生)的PDAC中,发现了一种新的具有高代谢活性的CAF亚型——meCAF。在meCAF丰富的PDAC中CD8+T淋巴细胞和单核/巨噬细胞的比例明显增加,化疗药物的细胞毒作用明显增强,并有较好的免疫治疗反应[41]。基于CAF的异质性和可塑性,更深层次的理解和识别与治疗耐药有关的CAF亚群及其背后的分子机制,将有利于找到新的治疗策略,特异性地针对CAF来克服胰腺癌治疗的耐药性。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:闫毅负责拟定写作思路,检索文献,撰写论文;郭亚荣负责课题设计;贾军梅负责指导、修改论文并最终定稿。
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
保健医苑(2022年6期)2022-07-08
中国临床医学影像杂志(2022年2期)2022-05-25
昆明医科大学学报(2022年4期)2022-05-23
中国医药科学(2022年5期)2022-05-05
临床肝胆病杂志(2021年10期)2021-12-22
抗癌(2021年4期)2021-12-01
天津医科大学学报(2021年4期)2021-08-21
奥秘(创新大赛)(2019年9期)2019-10-09
奥秘(2017年5期)2017-07-05
