
酸调节锆基 MOF 材料在光催化还原 Cr(VI)的应用
2022-05-13 05:38强涛涛尉梦笛
皮革科学与工程 2022年3期
强涛涛 ,尉梦笛
(陕西科技大学,陕西 西安 710021)
引言
工业的快速发展导致重金属离子铬的污染日益严重,其广泛存在于电镀、印染、制革等工业生产过程的废水中[1]。目前对于 Cr(VI)的处理技术包括离子 交换法[2]、膜 分 离技 术[3]、化 学 沉 淀 法[4]等 ,但 存 在能耗高,二次污染严重等缺陷。光催化技术可以利用 光生电子的还原 能 力,将 高毒性 的 Cr (VI)还 原 为低毒性的 Cr(III),能耗低且无二次污染,被认为是最有前途的处理技术之一[5-8]。
金属有机框架材料(MOF s)由于其高孔隙率、大比 表 面 积 以 及 可 调 控 的 结 构 等 突 出 优 势 而 备 受 关注[9-11]。现有的提高 MOF s 光催化活性的策略主要集中在以下几个方面:(1) 通过功能化有机配体来改变材料的光学性质,进而提升材料对光的响应[12];(2)通过与无机半导体材料复合形成异质结构改善MOFs 光催化性能[13];(3)将 MOFs 材料作为模板合成复合型光催化材料,提高传统光催化材料的催化性能[14]。此外,研究发现用于光催化还原的稳定MOFs 材料大都由含锆氧簇、铝氧簇等金属团簇与羧酸配体配位构成,导致高度晶化的 MOFs 缺少催化活性位点,光生电子 - 空穴对的复合率高[15-17],而利用单羧酸的竞争配位对 MOFs 进行缺陷调控是提高其光催化活性的另一种途径,但是很少有人探索。
选择高稳定性的锆基金属有机框架材料(UiO-66)为基底,利用三氟乙酸调节剂对 UiO-66进行缺陷结构改性,并将其应用于光催化还原处理Cr(VI)。
1 实验部分
1.1 主要材料与仪器
四 氯 化 锆 (ZrCl4),2- 氨基对苯二甲酸(NH2-BDC),三氟乙酸(TFA),上海麦克林生化科技有限公司 ;N,N-二甲基甲酰胺 (DMF), 甲 醇(MeOH),天津市天力化学试剂有限公司;重铬酸钾(K2Cr2O7),天津市北联精细化学品开发有限公司。试剂均为分析纯。
S4800 场发射扫描电镜,日本 Hitachi;D8 Advence X- 射线衍射仪,德国 Bruker;ASAP2460 比表面仪,美国麦克仪器;同步 TG-DSC 热分析仪,德国耐驰;X 光电子能谱仪,岛津;紫外 - 可见 -近红外分光光度计,美国安捷伦公;荧光光谱仪,英国爱丁堡。
1.2 UiO-66 的制备
参照文献采用简单的溶剂热法制备 UiO-66[18]。称取等物质的量比的 0.2331 g ZrCl4(1.0 mmol)和0.1812 g NH2-BDC (1.0 mmol) 超声溶解于 50 mL DMF 中,于 100 mL 高压反应釜内 120 ℃反应 24 h。冷却至室温后离心分离,并用 DMF 和 MeOH 各洗涤三次,经离处理后,将所得产物于 60 ℃真空干燥箱中干燥 12 h,得到淡黄色固体 UiO-66。
1.3 酸调节型 UiO-66 的制备
采用溶剂热法,通过添加酸调节剂制备缺陷型UiO-66。称取 0.2331 g ZrCl4(1.0 mmol)和 0.1812 g NH2-BDC(1.0 mmol)超声溶解于 50 mL DMF 中,再加入一定量的三氟乙酸,其中 TFA 的用量分别为0.5 mL(6.7 mmol)、1.0 mL(13.5 mmol)、1.5 mL(20.2 mmol)、2.0 mL(26.9 mmol),搅拌均匀后转移至 100 mL 高压反应釜内 120 ℃保持 24 h。所得产物处理方式同 UiO-66,干燥后样品记为 UiO-66,DU-0.5,DU-1.0,DU-1.5,DU-2.0。
1.4 光催化实验
将 0.02 g 催化剂超声分散于 40 mL,50 mg·L-1的 Cr(VI)溶液中暗反应 30 min,达到吸附 - 解吸平衡。以 300 W 氙灯模拟太阳光,使用 λ≥420 nm 的滤波片去除紫外光进行光反应,每隔 10 min 取反应悬浮液 2 mL,并用 0.22 μm 滤膜滤除光催化剂。采用国标法二苯碳酰二肼(DCP)对 Cr(VI)进行显色及λ=540 nm 处的吸光度[19],计算 Cr(VI)的相对质量浓度 ρt/ρ0,分析光催化活性。
2 结果与讨论
2.1 SEM 表征分析
图1是 UiO-66,DU-0.5,DU-1.0,DU-1.5,DU-2.0 的 SEM 图。图 1(a)可以看到,纯 UiO-66 由纳米颗粒团聚在一起,影响其在水中的分散性,导致与催化底物的接触面积减小,进而影响其催化性能。图 1(b)和 1(c)分别是添加 0.5 mL 和 1.0 mL TFA 的UiO-66(DU-0.5 和 DU-1.0)的 SEM 图,纳米粒子增大,团聚状程度相对减弱;图 1(d)是添加 1.5 mL TFA的 UiO-66(DU-1.5),纳米粒子出现八面体形貌,但整体形貌并不均匀;图 1(e)为添加 0.2 mL TFA 的UiO-66(DU-2.0),纳米粒子的形貌呈八面体形状。随着 TFA 添加量的增加,UiO-66 的分散性相对增强,颗粒尺寸逐渐变大,结晶度变高。

图1 (a)UiO-66;(b)DU-0.5;(c)DU-1.0;(d)DU-1.5 和(e)DU-2.0 的 SEM 图Fig.1 The SEM images of (a)UiO-66; (b)DU-0.5; (c)DU-1.0; (d)DU-1.5 and (e)DU-2.0
2.2 XRD 表征分析
图2为 UiO-66 和缺陷 DU-x 的 XRD 图谱。在2θ =7.43° 、8.54° 和 25.7° 处有特征峰 ,分别代表UiO-66 的(111)、(002)和(244)晶面,这与文献报道的 UiO-66 特征峰位置一致[20],表明催化剂成功合成。尖锐的 XRD 峰表明所合成样品具有较高的结晶度。经酸调节的 DU-x 都显示与 UiO-66 相似的 XRD 谱,没有明显的添加峰,这表明 DU-x 的结晶度在经过三氟乙酸调节后仍保持良好。此外,随着三氟乙酸用量的增加,其 XRD 特征峰逐渐增强,这种变化可以归因于三氟乙酸与母液中的配体 NH2-BDC 发生了竞争配位,这种竞争配位打破了配体与金属团簇之间原有的配位平衡,导致UiO-66 的结晶速度变慢,从而提高了样品的结晶度。
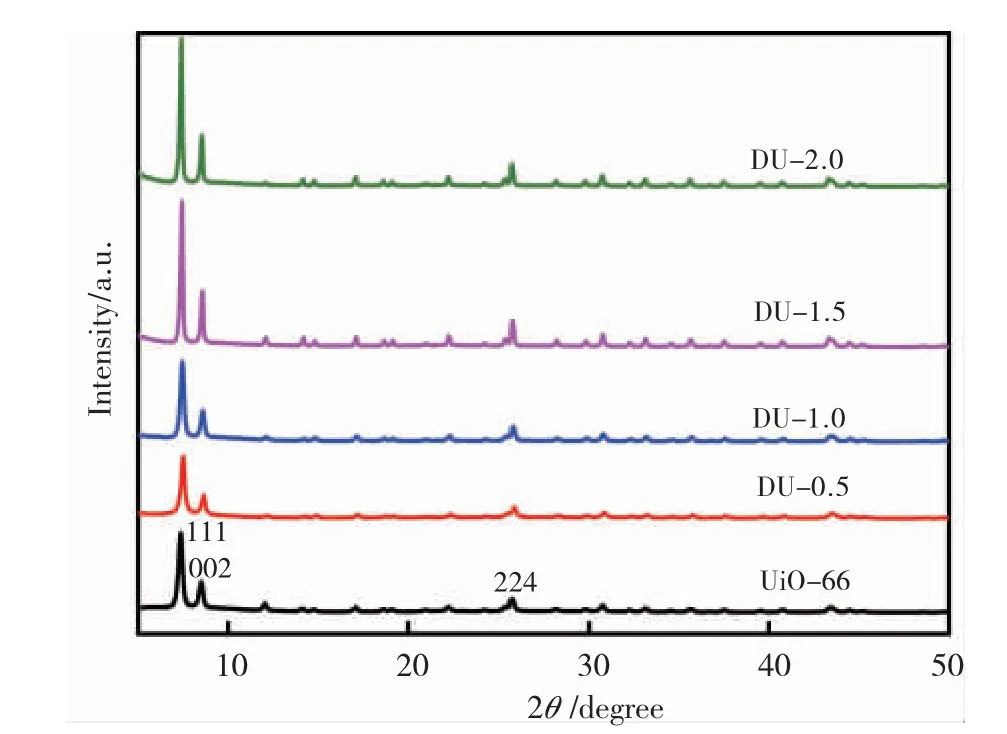
图2 UiO-66;DU-0.5;DU-1.0;DU-1.5 和 DU-2.0 的 XRD 图Fig.2 The XRD images of UiO-66; DU-0.5; DU-1.0;DU-1.5 and DU-2.0
2.3 BET 表征分析
图3为 UiO-66 和 添 加 1.5 mL TFA 后 样 品DU-1.5 的 N2吸附 - 脱附等温线,可以看到,该吸附- 脱附呈现典型的 IV 型等温线[21]。经三氟乙酸调节后样品的比表面积增加,由原来的 666.1 cm2/g 增大为 849.6 cm2/g,可为光催化反应提供更多的吸附和催化活性位点,从而增强光催化活性。

图3 UiO-66 和 DU-1.5 的 N2 吸附 - 脱附等温曲线Fig.3 N2 adsorption-desorption isotherms of UiO-66 and DU-1.5
2.4 UV-Vis DRS 分析
图4为催化剂的紫外 - 可见漫反射谱图。图中可知样品在 200 nm 和 291 nm 处均有吸收峰,对应锆氧簇的紫外吸收和有机配体的吸收峰[22]。UiO-66 的吸收边大约在 436 nm 处,DU-1.5 的吸收边大约在 464 nm 处,可以看到经三氟酸调节后,DU-x 的吸收边缘逐渐向较长的波长转移,表明缺陷位点的存在有利于增强材料对光的吸收性。
基于 UV-Vis DRS 光谱,用 Tauc plot 法[23]对其带隙进行计算,如图 4 中的插图所示,得出 UiO-66和 DU-1.5 的禁带宽度分别为 2.81 eV 和 2.68 eV,在相同情况下,DU-1.5 可以吸收更多的光子,产生更多的光生电子,具有更好的光催化性能。

图4 UiO-66;DU-0.5;DU-1.0;DU-1.5 和 DU-2.0 的紫外 - 可见漫反射光谱Fig.4 The UV-vis diffuse reflectance spectra of UiO-66; DU-0.5; DU-1.0; DU-1.5 and DU-2.0
2.5 光催化性能分析
图 5(a)所示为不同催化剂的光催化活性图。图中可以看到,在可见光照射 60 min 后,酸调节型UiO-66 的 光 催 化 活 性 明 显 高 于 UiO-66, 其 中 ,DU-1.5 的光催化活性最强,其相对质量浓度下降到96.33%,约为 UiO-66 的 2.21 倍。图中还可以看到在暗反应处理 30 min 前,随 TFA 用量的增加,对 Cr(VI)的吸附量也逐渐增大,这是因为经 TFA 调节后材料的比表面积增大,可以使 DU-X 和 Cr(VI)更有效的结合在一起,从而进一步提升催化剂对 Cr(VI)的光催化还原能力。利用一阶动力学方程评价样品的光催化还原速率(公式 1):

式中,ρ0为铬液的初始质量浓度,ρt为反应 t 时刻 Cr(VI)的质量浓度,单位:mg/L。k 为反应速率常数,单位:min-1。图 5(b)可以看到,DU-1.5 的反应速率常数 k(0.0282)约为 UiO-66(0.0028)的 10.07 倍,这表明三氟乙 酸的缺陷调控更有利于光生电子从配体到金属簇的迁移,增强材料的光催化活性。

图5 UiO-66;DU-0.5;DU-1.0;DU-1.5 和 DU-2.0 的光催化性能(a)以及(b)动力学曲线Fig.5 Photocatalytic performance (a) and (b) kinetic curves of UiO-66; DU-0.5; DU-1.0; DU-1.5 and DU-2.0
2.6 光催化机理分析
通过自由基捕获实验探究光催化反应机理[24]。如图 6 可知,加入 h+捕获剂 EDTA-2Na 后,Cr(VI)的光催化还原速率由原来的 91%上升到 94%,加入 e-捕获剂 KBr 后,光催化还原率直接降为7.5%,表明 e-的连续转移将 Cr(VI)还原为 Cr(III);此外,加入·O2-捕获剂 BQ 后,其还原率也受到了一定抑制,表明·O2-也是还原过程的活性物种。e-和·O2-在光催化过程中均起到还原作用。加入EDTA-2Na 后,样品的光催化效果增强,这归因于EDTA-2Na 与价带的 h+发生反应促进了光生电荷的分离。

图6 自由基捕获剂对 Cr(VI)还原率的影响Fig.6 Effect of free radical trapping agent on reduction rate of Cr(VI)
3 结论
(1) 经三氟乙酸调节剂改性后 UiO-66 的比表面积为 849.6 cm2/g,为 Cr(VI)的光催化还原提供了更多的吸附和催化位点。
(2)当三氟乙酸的用量为 1.5 mL 时,在可见光下表现 良 好 的 光 催化活性, 其光催化还原率是UiO-66 的 2.21 倍。
(3)e-和·O2-是光催化还原 Cr(VI)的主要活性基团,UiO-66 经三氟乙酸调节后,表现更高的电子- 空穴分离率。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
分子催化(2022年3期)2022-08-13
疯狂英语·新阅版(2021年9期)2021-10-30
电脑报(2020年40期)2020-11-06
中国科技纵横(2019年3期)2019-03-25
电脑知识与技术(2018年19期)2018-11-01
上海医药(2018年15期)2018-09-03
分析化学(2017年12期)2017-12-25
科技创新导报(2016年30期)2017-03-15
