
膦配体电子和空间效应对钯催化羰化酯化反应的影响
2022-08-13 06:56:46刘梦力夏春谷
分子催化 2022年3期
刘梦力, 曾 波, 胡 波, 李 臻*, 夏春谷
(1. 中国科学院兰州化学物理研究所 羰基合成与选择氧化国家重点实验室, 甘肃 兰州 730000;2. 中国科学院大学, 北京 100049; 3. 江苏华昌化工股份有限公司, 江苏 张家港 215634)
羰基(-CO-)是一类在有机合成领域用途广泛的官能团, 而羰基化反应是在有机分子中引入羰基实现增氧增碳的重要反应, 在精细和大宗化学品的工业生产, 以及有机合成中广泛应用. 在这类反应中,过渡金属催化的烷氧羰基化反应, 也称为氢酯化反应或羰化酯化反应, 是将不饱和化合物、 CO和醇转化为相应的酯的一种直接方法, 具有高原子经济性和环境友好性.
钯(Pd)配合物催化剂在羰化酯化反应中表现出较高的活性和通用性, 因此受到广泛的关注. 其中,具有不同电子和空间性质的膦配体不仅可以稳定中心金属, 也是影响催化剂反应活性和选择性的一个重要因素. 近几十年来, 研究人员设计合成出数量众多的新型膦配体, 使Pd-P催化体系的综合性能不断提高. 我们将重点介绍和评述Pd催化的烯烃羰化酯化反应中膦配体电子和空间效应对活性和选择性的调控作用, 希望对合理设计新型膦配体有所启迪,并对未来羰化酯化反应的发展方向进行了展望.
1 膦配体
膦配体(PR3)中磷原子的孤对电子可与中心金属原子配位形成配位键(σ供体), 而填充电子的金属3d轨道与膦配体σ*反键轨道可形成反馈键(π受体), 因此膦配体兼具σ-给电子能力和π-受电子能力(图 1), 其电子性质可以在很大的范围内调节,例如, 从“强σ-电子给体/弱π-电子受体”到“弱σ-电子给体/强π-电子受体”. 同时配位原子的σ-给电子能力和π-接受反馈电子能力进一步调控了金属中心的电子性质以及影响了对位的配位原子与金属中心的配位强度. P原子上取代基的改变使膦配体的空间位阻可以在很大的范围内变化, 进而影响到金属中心的配位数和其它配体(包括底物)的配位排布. 因此, 配体的电子性质与空间立体性质协同, 综合地影响了催化反应的各个步骤, 对过渡金属能高效地催化羰化反应的进行起到了关键作用[1]. 由此可见, 通过对配体的筛选, 改变金属中心的电子性质/空间位阻, 来优化催化中心的配位环境, 是调控Pd配合物催化活性以及反应化学/区域选择性的重要手段.

图1 膦配体电子性质示意图[2]Fig.1 Schematic diagram of the electronic properties of phosphine ligands[2]
膦配体的类型和结构具有多样化的特点, 包括: 单齿、 双齿和多齿膦配体; 非手性或手性膦配体; 以及一些特殊空间和电子结构的膦配体, 例如,hemilabile(半稳定)配体(具有一个强配位基团和一个弱配位基团, 呈现“半稳定性”)和笼状膦配体.图2显示了部分代表性膦配体结构.

图2 代表性膦配体结构Fig.2 Structures of representative phosphine ligands
2 膦配体电子和空间效应
膦配体修饰的钯催化剂与酸性助剂组成了不饱和化合物选择性羰化酯化的高效催化体系. 研究发现, 羰化酯化反应的活性、 化学选择性和区域选择性与膦配体的性质高度关联, 配体骨架结构和磷原子上取代基的不同引起配体电子性能和空间立体环境的改变. 在1970年以前, 配体结构上的几乎所有现象都用电子效应来解释, 直到Tolman系统研究了单膦配体的“配体效应”, 提出了电子效应和空间效应的定量方法, 才首创了有机金属配合物均相催化领域中对配体性质的系统性描述[3]. 此后大量文献证实膦配体空间效应和电子效应并非孤立地对金属配合物的活性、 稳定性和选择性产生影响, 两者之间相互作用, 具有协同性[4].
根据Tolman的量化方法, 单齿膦配体PR3的给电子能力可以通过测量化合物Ni(CO)3(PR3)的红外羰基伸缩振动吸收峰νCO来表征[5](图3). 为定量描述R基团对PR3配体给电子能力的影响, 引入电性参数χi, 则:
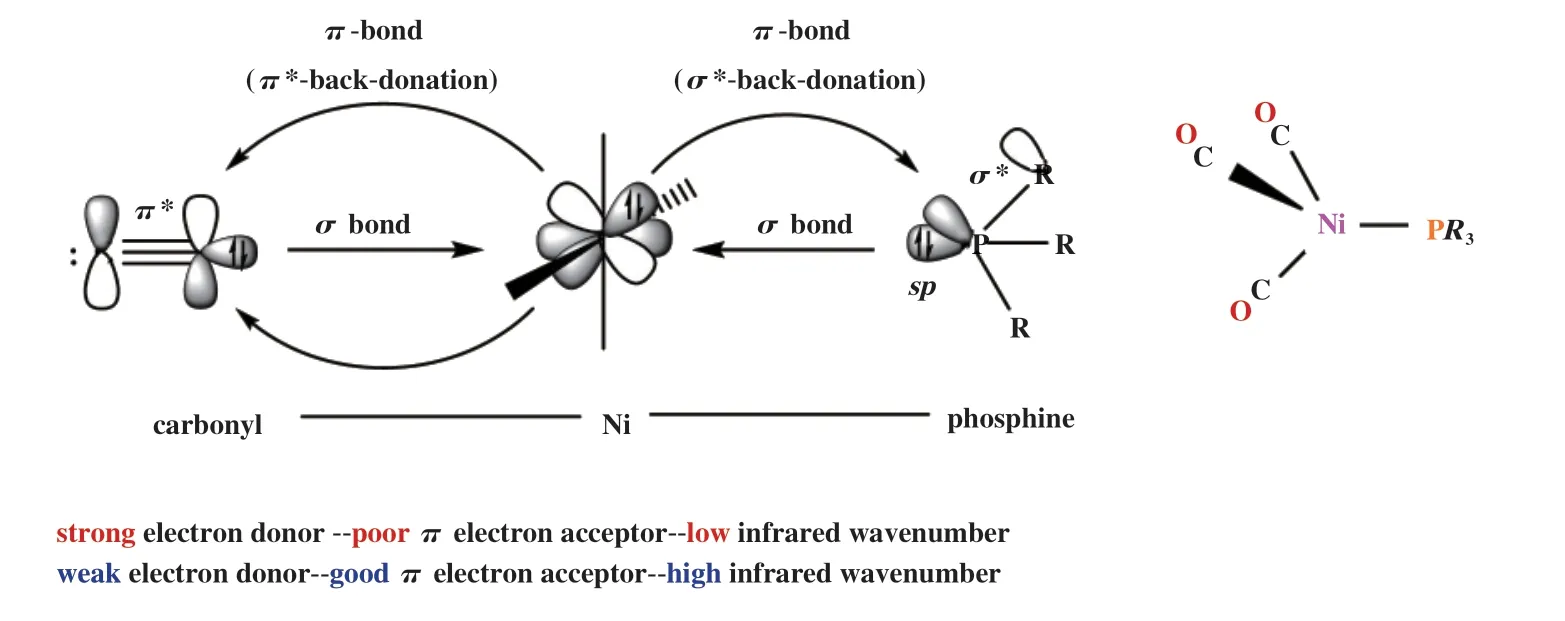
图3 PR3给电子能力的量化[5]Fig.3 Quantification of electron donating capability of PR3[5]

表1中列出了代表性R基团的χi值.

表1 膦配体R 取代基的χi值[5]Table 1 χi for R substituents of phosphine ligands[5]
PR3的空间效应则用锥角θ来近似衡量[3-6](图4 a), 锥角越大, 配体空间位阻越大, 即空间立体效应越显著. 锥角最初是以对称单齿膦配体与镍中心以四面体形式排列而提出的, 对于不对称膦配体,锥角可以通过取代基之间的三个角度的平均值来估计. 由于在Tolman最初的研究中, 配体被折叠成尽可能小的锥体, 而没有考虑其他可能结构的相对稳定性, 因此, 最初的方法适用于小的配体, 如PH3或PMe3, 当它应用于更复杂的单齿或双齿配体时, 其有效性大大降低. 为克服这些缺陷, 化学家又发展了立体角Ω[7-8](配体原子投射到以中心金属原子为中心的球体上的阴影面积)、 掩埋体积百分比%Vbur[9-11](任何给定的配体在一个以金属原子为中心的半径为0.35 nm的球体内所占的体积, 最初用于N-杂环卡宾(NHC)配体, 现在也应用于单、 双齿膦配体)等描述符来描述膦配体的不同立体环境和电子性质(图4 b和c).
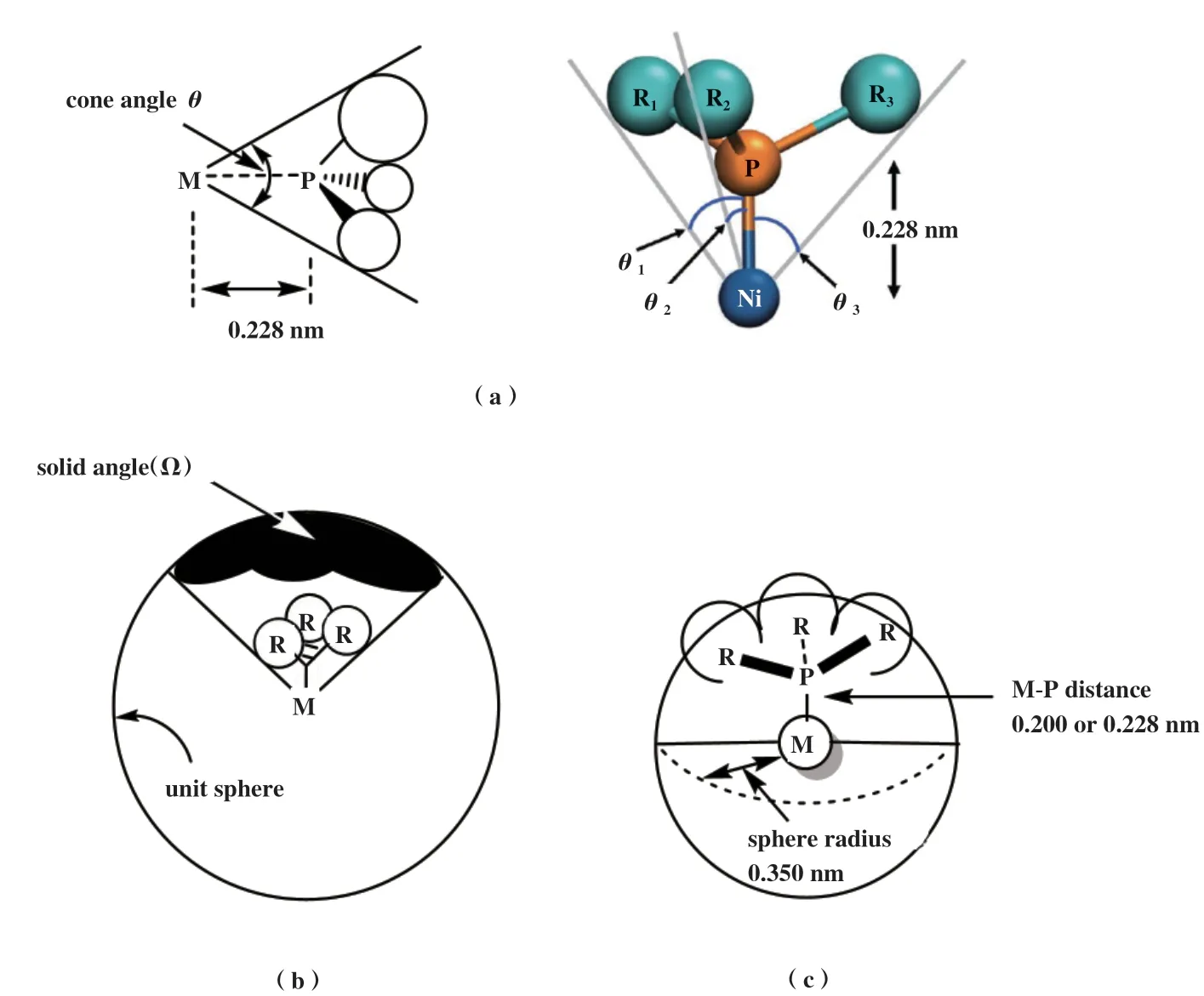
图4 用于衡量单齿膦配体的空间效应的描述符(a: 锥角θ[5]; b: 立体角Ω[7]; c: 掩埋体积百分比%V)Fig.4 Descriptors for measuring the steric effect of monodentate phosphine ligands(a: cone angle θ[5]; b: solid angle Ω[7]; c: the buried volume %V)
Tolman空间参数和电子参数是定量和定性理解配体对过渡金属配合物性质影响的有力工具. 这些配体参数对金属-配体和配体-底物相互作用的影响是催化剂合理设计的基础. 膦配体对M-P键和反应活性的空间贡献和电子贡献的定量研究, 在很大程度上促进了新型过渡金属配合物催化活性的发现和提高.
不饱和化合物羰化反应的区域选择性调控对于直链或支链羰化产物的定向制备具有重要意义. 众所周知, 对于末端烯烃、 二烯烃或末端炔烃的羰化反应, 受热力学因素的限制, 产物主要为支链的羰基化合物[12-15]. 研究发现, 相对于单齿膦配体, 双齿膦配体更倾向于生成直链羰化产物. 在羰化酯化反应中, 双齿膦配体的应用十分广泛, 其双齿螯合作用提高了催化剂稳定性, 并且能提高反应区域和立体选择性. 但是Tolman的电子和空间参数很难适用于双齿膦配体, 原因在于双齿膦配体骨架两个磷原子之间的距离是一个重要的配体参数.
除了前面提到的立体角Ω和掩埋体积百分比%Vbur, 用于描述双齿膦配体的参数还包括口袋角(pocket angle)[16]、 排斥能(repulsive engergy)[17]、 可及分子表面(acessible Molecular Surface, AMS)[18]和自然咬角(natural bite angle). 其中, 自然咬角很容易通过简单的分子力学模型计算得到, 也是双齿膦配体中应用最广泛的参数. 1990年, Casey等[19]在研究铑催化剂与双齿膦配体的几何构型对反应选择性的影响时, 提出了“咬角(bite angle)”的概念(见图5). 自然咬角(βn)是指当配体-金属的张力能为0时,配体-金属-配体(P-M-P)形成夹角的计算值. Casey认为双齿膦配体具有较大的βn(接近120°), 同时有较好的柔韧性, 将有利于提高正构醛的区域选择性.
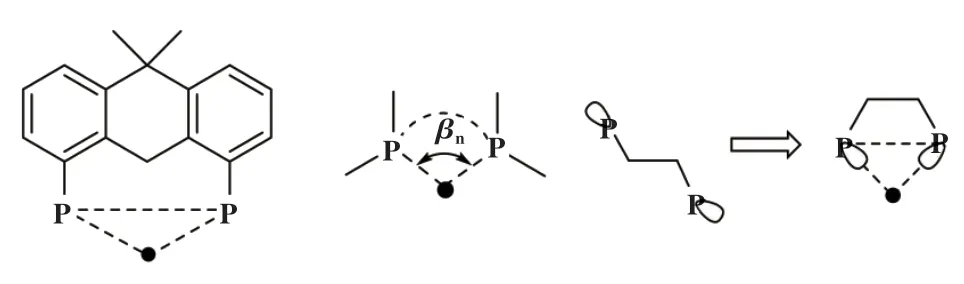
图5 双齿膦配体自然咬角示意图[20]Fig.5 Bite angle of bidentate diphosphine ligand[20]
配体咬角的改变通常会对空间结构产生两种效果, 其一是具有较宽咬角的配体对空间的要求更高,将对金属中心的其他配体施加更大的空间排斥; 其二是固定的配体咬角所具有的电子或轨道效应可将金属中心推向首选的几何形状.
通过增加连接两个P原子之间的链长可以很容易地改变螯合双膦的咬角. 1,2-双(二苯基膦基)乙烷 (dppe, L6)与金属配位形成非常稳定的五元环螯合物, 当碳链长度达到 C4后, 例如1,4-双(二苯基膦基)丁烷(dppb, L8), 配体具有相当大的灵活性, 并且螯合能力降低, 有利于形成桥连双金属结构, 从而导致不同的反应途径和机制. 为了开发有用的、 更大咬角的配体, 需要更刚性的结构, 例如含有芳环骨架的Xantphos(L16)和BINAP(L17).
3 钯配合物催化羰化酯化反应
不饱和化合物与CO的催化羰基化在形成 CC 键的同时将羰基官能团引入到底物分子中, 从而得到酯、 酸、 醛、 酮等大宗基础化学品. 其中, 在过渡金属配合物催化剂作用下, 烯烃、 炔烃、 联烯等不饱和化合物的羰化酯化反应(Reppe羰化反应)因产物酯具有稳定性好、 应用面广、 需求量大等优点,得到广泛研究(图6). 虽然Ru[21-24]、 Rh[25]、 Co[26-29]和Ni[30]的配合物都可以催化羰化酯化反应进行,但是钯配合物催化剂的研究最为广泛也最具吸引力[31-34].

图6 不饱和化合物的羰化反应Fig.6 Carbonylation of unsaturated compounds
自从二十世纪六十年代, 钯催化 Reppe羰化反应被首次报道以来[35-36], 由于钯催化剂与配体的相互作用能够更好地促进和调控羰化反应的进行, 使得钯催化体系得到迅猛发展[37-38]. 在钯催化不饱和化合物的羰化酯化反应中, 配体不仅能稳定各自的金属中心, 而且能从根本上改变催化剂体系的选择性和活性[39-40].
Pd催化羰化酯化反应的实验[41-44]和理论[45]研究表明, 反应机理为金属氢化物([Pd-H]+)途径(图7). 以烯烃为例, 首先, 钯前体在酸助剂作用下生成钯氢化物活性物种, 随后烯烃双键与[Pd-H]+配位加成形成钯烷基中间体, 接着CO配位、 迁移插入Pd-C 键得到钯酰基化合物, 最后, 亲核试剂醇进攻钯酰基化合物得到酯化产物, 同时释放[Pd-H]+物种进入下一次循环. 在烯烃的羰化酯化反应中,由于烯烃插入方式不同以及双键异构化, 会导致反应同时生成支链(支化)和直链(线性)酯产物. 一般来说, 对于末端烯烃的羰化酯化, 使用单齿膦配体主要生成支链产物, 而使用双齿膦配体直链酯选择性显著增加, 尤其是在使用大位阻螯合双齿膦配体的情况下, 直链产物选择性可达99.9%. 因此, 深入了解配体性质与催化性能之间的关系, 将使我们能够通过合理设计配体, 优化电子和空间效应来获得更多性能优异的催化剂.

图7 钯催化烯烃羰化酯化反应的一般机理Fig.7 General catalytic pathway of palladium-catalyzed carbonylation of alkenes
3.1 钯-单齿膦配体催化体系
早期钯催化烯烃的羰化酯化反应使用无膦配体催化体系(如PdCl2/HCl[46]、 PdCl2/CuCl2[47-48]), 反应条件不温和, 容易产生钯黑沉积导致催化剂失活,需要加入化学计量的氧化剂将Pd(0)再次氧化成Pd(II). Cavinato等[49]采用[PdCl2(PPh3)2]-PPh3体系催化3-丁烯乙酯的羰化酯化, 发现在甲苯或 2-丁酮溶剂中, 产物以支链酯(甲基丁二酸二乙酯)为主, 但是支链产物的选择性随PPh3浓度增加而下降. 作者认为可以用空间效应进行解释, 即在高浓度的PPh3存在下, 几种催化活性物质之间的平衡会向体积较大、 活性较低的中间体转移, 使直链产物选择性升高, 而产率降低.
使用弱电子给体单齿膦配体可以提高支链产物的区域选择性, 如图8所示多氟烷基取代苯基膦配体修饰的钯配合物[Pd(OAc)2(PAr3F)2], 在苯乙烯甲氧基羰基化反应中, 虽然反应速率不及[Pd(OAc)2-(PPh3)2], 但是, 支链产物2-苯基丙酸酯的选择性明显高于[Pd(OAc)2(PPh3)2]体系[50]. 通过对比[Pd(OAc)2(PAr3F)2]和[Pd(OAc)2(PPh3)2]的31P{1H}NMR-谱发现, 溶液中[Pd(OAc)2(PAr3F)2]由顺式和反式异构体组成, 而[Pd(OAc)2(PPh3)2]则全部为反式结构. 这种含氟配合物容易形成顺式异构体的趋势在催化过程中发挥了有益的作用. 作者认为含氟配体诱导的空间较少, 倾向于形成更多的顺式(膦)钯配合物中间体, 这可能是 [Pd(OAc)2(PAr3F)2]催化剂体系中异正比较高的原因.
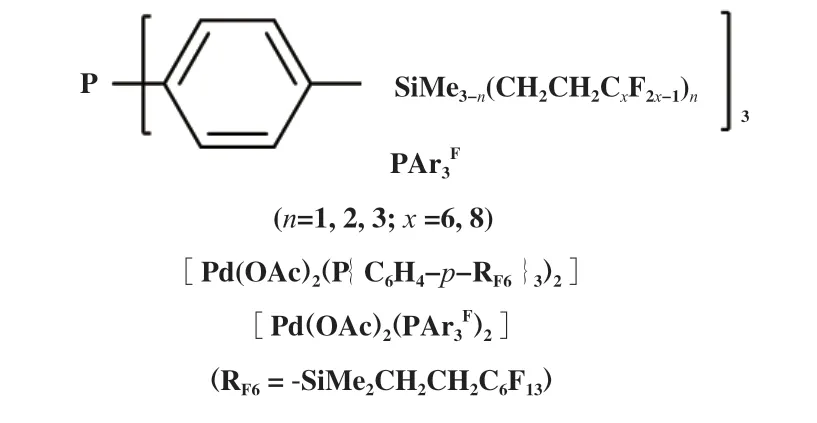
图8 含氟单齿膦配体及其钯配合物[50]Fig.8 The fluorous phosphine ligand and palladium complexe[50]
通过合理选择金属前驱体和添加配体, 可以对羰化反应的区域选择性进行调控. del Rı́o等[51]详细研究了钯催化苯乙烯氢羧基化反应中单齿膦和双齿膦配体对区域选择性的调控, 发现锥角θ>145°的单齿膦配体与[PdCl2(PhCN)2]组成的催化剂对支链产物有高的区域选择性. 作者认为无论单齿膦体系还是双齿膦体系都经历了Pd-H循环,并且由于空间和电子的原因, 单齿膦配体采取反式配位取向(图9, 1). 由于关键中间体2只含有一个膦配体, 且空间位阻很小, 因此趋向于形成稳定的η3-苄基钯中间体3, 经CO配位, 迁移插入得到支链产物(图9).

图9 tran-配位中间体向η3-苄基钯中间体转变[51]Fig.9 The transformation of tran- coordination species to η3-benzyl palladium intermediate[51]
为了更深入地了解控制区域选择性的机制, del Rı́o等[52]利用原位高压核磁实验评估了膦配体和钯前驱体对苯乙烯羰化区域选择性的影响. 作者认为单齿膦配体采取反式配位而双齿膦配体采取顺式配位是区域选择性控制的关键. 在顺式双齿螯合配合物中, 配位于钯中心的膦配体取代基与苯乙烯的芳基之间有很强的相互作用, 容易形成直链烷基-钯物种. 而且双齿膦钯配合物随着螯合环增大其稳定性降低, 导致一个Pd-P键断裂, 形成反式单膦配位, 支链产物选择性增大, 出现和单齿膦配体相类似的结果. 此外, 动力学研究表明, Pd(0)配合物、 钯氢物种、 钯烷基物种和钯酰基物种之间存在反应平衡, 因此可逆性似乎对于区域选择性也起着重要作用. 支链钯酰基中间体比线性钯酰基物种更稳定,但是后者与亲核试剂的反应更快, 因此区域选择性在Curtin-Hammett规则下很难预测.
通常, 单齿膦配体-钯催化剂体系需要膦配体过量, 以保证催化剂的稳定. 为了探究单齿膦-钯配合物能否构建性能优于双膦-钯配合物的催化体系, Fuentes等[53]研究了金刚烷基笼型单齿膦配体CYTOP 292(图2, L22)在烯烃羰化酯化反应中的应用. 研究发现膦钯比为1的配合物催化剂[Pd(Cl)-(L22)(η3-C3H5)]在低温、 低催化剂用量下具有优异的活性和支链产物选择性. 该配合物在苯乙烯与乙醇的羰化酯化反应中的催化效果远好于[Pd(Cl)-(PPh3)(η3-C3H5)], 并且亲核试剂可以扩展到异丁醇、苄醇和叔丁醇(图10).
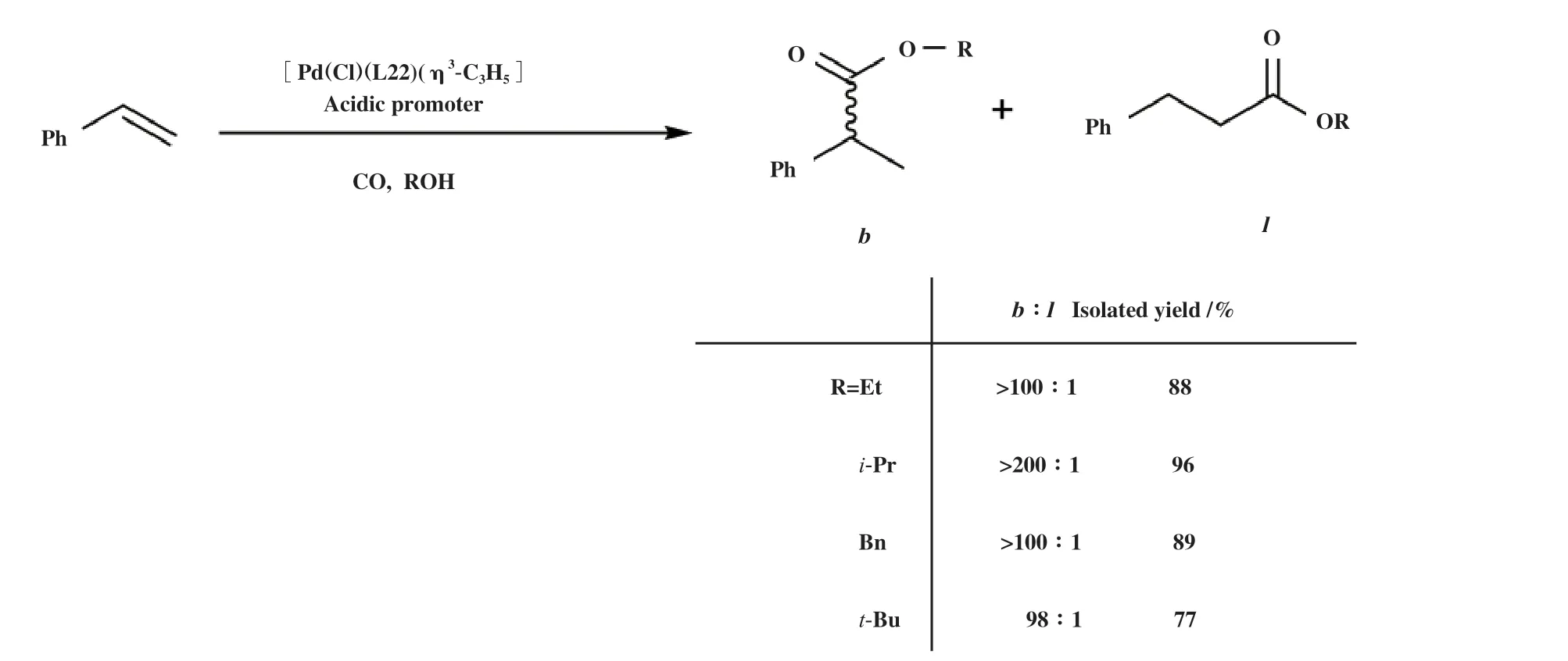
图10 [Pd(Cl)(L22)(η3-C3H5)]催化苯乙烯羰化酯化[53]Fig.10 Alkoxycarbonylation of styrene catalyzed by [Pd(Cl)(L22)(η3-C3H5)] [53]
尽管大多数研究表明单齿膦有利于支链产物的形成, 但是研究者通过对配体、 助剂、 反应条件等参数的调控实现了单齿膦-钯配合物催化直链酯的选择性合成. Knifton[54]研究了钯-单齿膦配合物与第四主族金属卤化物组成的催化剂体系([(p-XC6H4)3P]2-MCl2, X=H、 CH3、 CH3O、 Cl; M= Sn或Ge)对1-庚烯的羰化酯化反应, 直链酯产物选择性达到85%~89%(摩尔分数). 通过比较发现, 对于一系列锥角相似的芳基单齿膦配体, 只有碱性适中的配体可以增强钯氢物种HPd(SnCl3)(PPh3)L的活性, 促进烯烃对Pd-H的反-马氏加成, 最终得到直链酯产物. 这是由于大体积膦配体和具有强π-电子接收能力的SnCl3-协同作用产生了具有特殊空间位阻效应的催化剂体系. 这种空间约束力既有利于反-马氏Pd-H加成, 又有助于提高空间位阻较小的σ-烷基钯和σ-烷酰基钯异构体的平衡浓度.
脂肪族长链烯烃羰化酯化反应的区域选择性调控存在一定挑战, 例如, 由于1-辛烯的异构化, 其羰化酯化反应会产生4种C9酯. Alper课题组[55]基于CYTOP 292 的优异性能, 以路易斯酸SnCl2或Ti(OiPr)4为添加剂, 开发了PdCl2/CYTOP 292催化体系,实现了脂肪族长链烯烃羰化酯化反应的区域选择性调控. 该体系能够催化脂肪族烯烃的反-马氏羰化酯化, 得到以直链为主的产物(选择性在 70%~96%),产率大于 93%(如图11所示). 作者通过对比不同膦配体的催化效果, 发现在相同的反应条件下, 只有PdCl2/P(p-MeO-Ph)3体系可与PdCl2/CYTOP 292相媲美, 转化率为100%, 直链产物选择性为93%. 显然,配体的性质是区域选择性的关键, 配体锥角和托尔曼电子参数(TEP)与反应活性和区域选择性相关[56].

图11 脂肪族烯烃的反-马氏羰化酯化反应[55]Fig.11 The anti-markovnikov alkoxycarbonylation of aliphatic alkenes[55]
长链脂肪族烯烃双键插入Pd-H键形成仲碳-钯中间体(马氏加成)的空间效应增加, 使羰基化反应中支链产物的选择性生成更具挑战性.2016 年, Li等[57]探索了钯和一系列单齿膦配体催化长链脂肪族烯烃的马氏羰化酯化反应. 研究表明, 配体空间位阻的增加有利于形成所需的支链产物, 特别是联芳基骨架单齿膦配体性能突出, 其中, PdCl2/N-苯基吡咯膦型催化体系具有最佳活性和区域选择性. 如表2 所示, 各种脂肪族烯烃包括功能化烯烃生成支链产物的产率为71%~97%, 区域选择性为49%~99%. 进一步, 作者通过DFT理论计算证明, 支链产物的高区域选择性主要由相应羰基配位烷基钯中间体[L2Pd(CO)(isopropyl)]+和[L2Pd(CO)(propyl)]+在热力学稳定性上的差异决定.

表2 钯催化烯烃反-马氏羰化酯化[57]Table 2 The anti-markovnikov alkoxycarbonylation of olefins over palladium catalyst[57]
综上所述, 单齿膦配体-钯体系对区域选择性的调控在很多情况下需要钯前体、 配体、 助剂和溶剂的协同作用, 以获得对单一构型产物满意的产率和选择性. 单齿膦配体种类繁多, 对其电子和空间性质的研究和应用筛选工作量巨大, 随着数据积累、 计算水平及分析手段的不断发展, 基于大规模数据驱动的催化过程设计和优化方法有望成为基于有机膦配体的过渡金属催化研究快速发展的关键推动力之一[58].
近日, Gensch及合作者[59]开发了一个覆盖单齿有机膦配体的数据库kraken(图12). 利用量子力学方法, 计算了1558个单齿有机膦配体的描述符,并训练了机器学习模型来预测超过30万个新配体的性质. 利用kraken可以系统地探索有机膦配体的性质空间, 使基于有机膦配体的催化剂设计成为可能, 促进反应过程参数的优化, 激发新的配体选择, 并促进新的有机膦化合物的合成.

图12 单齿有机磷配体数据库kraken[59]Fig.12 Kraken: A comprehensive database of monodentate phosphorus ligands[59]
3.2 钯-双齿膦配体催化体系
双齿膦配体的螯合配位效应可以有效稳定金属中心, 并强烈影响金属中心的化学性质, 因此催化性能对配体环境的变化很敏感. 典型的双齿膦配体dppe(L6)在1959 年被报道[60], 随后, Cannel 等[61]将其用于钴催化的氢甲酰化反应, 但是与无膦羰基钴体系相比性能没有显著变化. 由于双齿螯合配位作用降低了催化循环中配体的解离倾向, 使得催化条件下的中间体结构更加明确, 近些年来, 研究人员对双齿膦配体在羰化反应中的重要作用进行了大量研究.
一般来说, 双齿膦配体的性质及其配合物的催化性能可以通过改变配体的主链及P原子上取代基来调变. 4,5-双二苯基膦-9,9-二甲基氧杂蒽(L16, Xantphos)配体最早由van Leeuwen等[62]设计合成, 具有特殊的富电子性和较大的空间位阻, 是羰基化反应最著名的双齿膦配体之一. 在调控Pb配合物催化活性和选择性方面,研究较多的是许多大位阻双齿膦配体, 如1,2-双(二叔丁基膦甲基)苯[63-64](L13, dtbpx)、 1,2-双(二叔丁基膦)苯[65-66](L12)、 1,3-双(二叔丁基膦)丙烷[67](L10, dtbpp)、 双(二苯基膦甲基)双环庚烷[68](L11)、 双(磷杂金刚烷基)烷烃[69-70](L23和L24)和双(二苯基膦基)茂金属[71].
双齿膦配体自然咬角的变化对催化选择性有重要影响[72-75], 例如, Freixa和van Leeuwen[76]对双齿膦配体咬角效应对铑催化氢甲酰化反应、 镍催化氢氰化反应和钯催化乙烯羰化合成聚酮或丙酸甲酯反应的影响进行详细研究, 并指出P-M-P咬角对3种反应的选择性和反应速率起着至关重要的作用. 为更加合理地解释咬角的影响, 双齿膦配体的咬角被分解为空间咬角效应和电子咬角效应以区分不同的作用方式. 分析认为, 氢甲酰化反应的区域选择性主要由空间因素决定, 而反应速率则由咬角的电子效应决定, 钯催化羰基化反应的选择性主要受位阻因素的影响.
双齿膦-钯配合物催化羰化酯化反应对咬角依赖关系的最具显示度的证据之一, 是CO/乙烯共聚和乙烯的羰化酯化之间的微妙平衡(图13).

图13 乙烯羰化酯化和共聚反应[77]Fig.13 Alkoxycarbonylation and copolymerization of ethene[77]
丙酸甲酯(MeP)是CO/乙烯共聚反应的最小可能产物, 由两个单体插入后立即发生链转移时产生.因此, 共聚和烷氧羰基化之间的选择性控制意味着在链增长和链转移之间进行调控, 这恰好能通过配体的修饰来实现. 最具活性和 MeP 选择性的配体是那些在P上具有大体积取代基和/或大咬角的配体,例如, 基于L10或L13的催化剂以大于99.9%的选择性产生MeP, 但基于L7或L14的催化剂则生成乙烯/CO共聚低聚物和聚酮, 这表明在影响催化剂羰化酯化效率方面, 配体空间效应比电子效应更重要.
实验研究和理论计算都表明双齿膦配位的Pd中心周围的空间拥挤程度是羰化酯化选择性和活性的决定性因素. 例如, Ahmad等[78]通过量化计算研究比较了dtbpx和dmpx[1,2-双(二甲基膦甲基)苯]的空间体积对Pd催化乙烯羰化酯化活性的影响, 结果表明含大位阻取代基的dtbpx破坏了最低能量中间体(MARI)的稳定性, 降低了最高能量过渡态(HETS)的势能, 从而降低了总反应的能垒, 提高了反应活性. 而dmpx由于空间体积减小, 稳定了MARI, 在一定程度上提高了HETS的势能, 从而增加了总反应能垒. 这些结果与实验观察完全一致,随着双齿膦配体体积的增加, 对酯化反应的选择性和反应速率也增加.
在不饱和植物油的异构/羰化酯化反应中, 通过对[(dtbpx)Pd(OTf)2]和[(dmpx)Pd(OTf)2]的对比研究, 也证明大位阻的dtbpx更倾向于形成直链Pd-烷基物种, 而相应直链Pd-酰基物种醇解的能垒也较低, 转化速率快[79]. van Leeuwen及其同事[76,80-81]在配体咬角对催化活性和选择性的影响方面做了很多研究. 利用QM/MM研究钯催化羰化反应中还原消除步骤, 结果表明, 宽咬角配体使反应物失稳, 使过渡态稳定, 加速了反应的进行, 从而将Pd-双齿膦配合物的咬角与反应速率关联起来.
Pd配合物催化芳基烯烃羰化酯化通常可获得较高的线性产物, 区域选择性与双齿膦的顺式配位方式以及P-Pd-P咬角之间存在相关性. 具有宽咬角的DPEphos(咬角102.7°)和Xantphos(咬角110°)在苯乙烯羰化反应中提供了高转化率和几乎全部的线性产物. 2004年, Bianchini等[82]报道了4种1,1’-双(二苯基膦)茂金属-Pd(II)催化剂在苯乙烯羰化酯化反应中的催化性能, 4种催化剂均得到线性产物(3-苯基丙酸甲酯区域选择性高达85%)(图14). 虽然线性区域选择性的原因仍然是一个有争议的问题, 但是作者认为P原子上苯基与支链烷基苯的相互作用所产生的空间位阻是形成直链型酯的决定因素[51,83]. 相比于催化剂1(咬角96.37°)和3(咬角97.47°), 含八个甲基的催化剂2 具有最大P-Pd-P咬角(101.3°), 因此在空间上4个苯环更靠近配位的烷基, 从而表现出最好的直链选择性.
除了咬角效应, 其他参数如电子效应和空间效应在决定区域选择性的步骤中所起的重要作用也必须考虑. 2006 年, van Leeuwen小组[84]报道了双齿膦配体电子效应对钯催化苯乙烯羰化酯化反应影响的研究, 提出双齿膦配体的电子效应能够控制苯乙烯羰化酯化反应的区域选择性, 使用缺电子的类 DPEphos双齿膦配体(图14)能够提高支链酯类产物的产率, 苯乙烯羰化酯化产物支链和直链比为74/26.

图14 1,1’-双(二苯基膦)茂金属-Pd(II)催化剂及类DPEphos配体结构[82,84]Fig.14 1,1’-Bis(phosphine)metallocenes-Pd(II) catalysts and DPEphos analogue[82,84]
最近, Tay等[85]合成了一系列芳基桥联的含Phobane基团的双齿膦配体(图15), 研究了这些配体对钯催化烯烃羰化反应的影响. 含Phobane基团的单齿膦配体PhobPR(烷基-9-膦杂双环壬烷[3.3.1])最早由Shell公司用于钴催化烯烃还原氢甲酰化直接制备洗涤剂醇[86]. Phobane及相关配体中C-PC桥基的应变力影响了孤对电子轨道的性质, 从而改变了最高占有轨道(HOMO)的能量, 并调节配位P原子的σ-给体和π-受体性质. 因此, 这类配体通常具有较弱的σ-电子给体和较好的π-电子受体能力. 由于Phobane的合成涉及到使用危险的PH3,因此, 有关此类配体的报道并不多.

图15 芳基桥联含Phobane双齿膦配体[85]Fig.15 Aryl bridging bidentate phosphine ligands containing Phobane[85]
Tay将图15 的配体用于钯催化1-辛烯在2-乙基己醇溶剂中的羰化反应, 发现不同的配体对产物选择性有显著的影响. 使用乙基桥联的配体BCOPE主要生成壬醇(95%选择性), 而芳基桥联的配体受芳环上取代基的影响, 产物选择性发生了较大变化. 其中含给电子取代基的配体(L30和L31)有利于发生还原氢甲酰化生成醇, 而含吸电子取代基的配体(L28)则有利于羰化酯化反应生成壬酸2-乙基己酯(73%选择性). 根据羰化酯化过程醇解步骤的机理(图16), 配位于Pd中心的醇失去质子后, 形成中间体1, 随后发生还原消除得到产物酯. 配体L28上CF3基团的吸电子效应使电子云密度远离P原子, 导致金属中心更加亲电, 从而极化配位醇分子的OH键, 促使H+脱除, 加速了醇解步骤的速率.
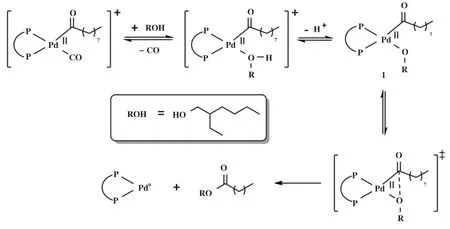
图16 羰化酯化过程醇解机理[85]Fig.16 Mechanism of alcoholysis process foralkoxycarbonylation[85]
内烯双键的反应性通常较低, 利用含螯合双齿膦配体的金属配合物催化性能对配位环境的变化较敏感的特性, 可以通过配体电子和空间性质来影响催化剂在内烯烃羰化酯化反应中的活性和选择性.例如,meso/rac-L24与Pd(OAc)2组成的催化剂在内十四烯羰化酯化反应中, 产物正异比达到78∶22,与α-十四烯结果相近, 但是反应速率仅为后者的0.4倍, 说明内烯异构是限速步骤, 线性产物选择性高是末端烯的羰基化速率快的结果[70]. 配体L10(dtbpp)和L24主链均为-(CH2)3-, 但L10-Pd(OAc)2几乎没有异构/羰基化反应的活性, 两者性能的差异与L24笼状结构特殊的电子和立体特性密切相关.通过测量化合物Ni(CO)2(diphos)的红外羰基伸缩振动吸收峰νCO来表征meso/rac-L24的σ-/π-电子性质, 当diphos =meso/rac-L24时, νCO为2002 cm-1,远高于L10(1976 cm-1), 也略高于L8(dppp, 1997 cm-1), 因此可以推断, L24配体的P原子电子性质与二芳基膦片段更相近, 属于弱的σ-供体/好的π-受体.
meso-L24-Pd 和rac-L24-Pd 在丙烯及C11/C12内烯羰化酯化反应中表现出不同的催化活性, 前者活性大约为后者的2倍, 再次证明螯合配位的钯催化剂结构的微妙变化也会对催化活性产生很大影响.
Behr等[87]在研究油酸甲酯羰化酯化制二酯类化合物的反应中也观察到配体的电子和空间性质对反应速率以及异构化过程的影响(图17). 以[Pd2-(dba)3]为催化剂前体, 不同的双齿膦配体显示出不同的反应结果, 其中dtbpx配体具有非常好的内烯烃异构/羰化酯化活性(转化率80%, b∶l=19∶81).Cole-Hamilton小组[64]也曾报道过在[Pd2(dba)3]-dtbpx催化体系下, 无论是末端烯烃还是内烯烃, 都能转化为末端直链酯类产物, 其中直链选择性达到98%, 说明该配体有利于内烯异构到端位, 而且仅对端位烯烃羰化酯化有促进作用. 使用电子效应相似、 咬角更大的L33, 则完全没有催化活性. 作者发现Xantphos(L16)表现出与L13相近的活性, 但产物选择性相反(转化率70%,b∶l=77∶23). 氧杂蒽骨架的Xantphos配体是一类具有蝴蝶型结构的大位阻配体, 配位原子可以很好地与许多过渡金属配合形成配合物, 咬角远大于90°, 因此Xantphos与Pd通常形成反式配位化合物[88], 而这种反式配位有利于内双键参与反应. 通过改变Xantphos骨架上C10位点的基团, 可以诱导咬角的微小变化, 使键合模式发生改变, 其催化活性随之发生巨大转变. 如使用图17中配体Nixantphos, L34(咬角114°), 反应转化率只有20%, 证明配体咬角的改变对催化活性有极大影响. 改变P原子上取代基, 调变催化剂的酸碱性,发现L35完全没有活性, 说明配体电子效应在催化循环中同样发挥了重要作用.
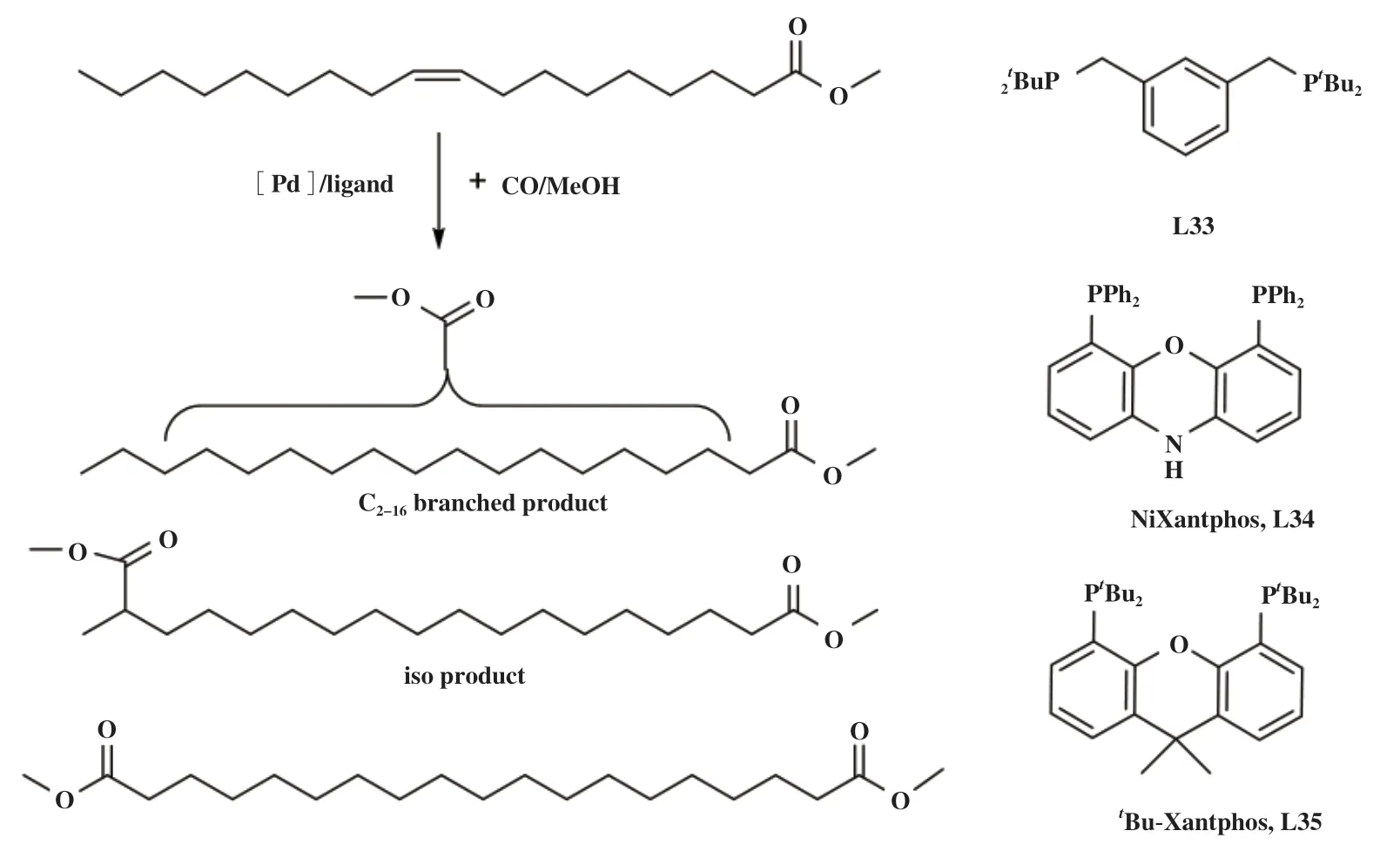
图17 油酸甲酯羰化酯化[87]Fig.17 Alkoxycarbonylation of methyl oleate[87]
在异构/羰化酯化反应中具有高区域选择性的双齿膦配体的共同特征是具备刚性的骨架、 相对宽的P -M- P咬角(~ 100°)和P原子上大体积叔烷取代基. 基于此, Nobbs等[89]设计合成了新型膦配体BPX(L18). BPX与Pd(OAc)2组成的催化体系能够很好地催化末端烯烃、 内烯烃、 官能化烯烃的羰化酯化反应, 高选择性生成末端酯, 表现出比Pd-dtbpx体系更优越的性能(图18).
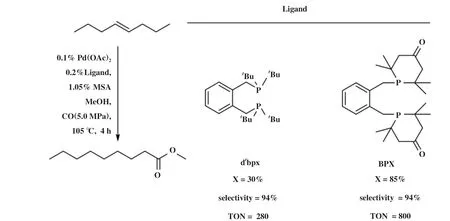
图18 BPX与dtbpx在tran-4-辛烯羰化酯化反应中的比较[90]Fig.18 Comparison of BPX and dtbpx in methoxycarbonylation of tran-4-octene[90]
BPX配体与dtbpx结构相似, 咬角接近, 不同的是BPX上的P原子被限制在一个含有吸电子羰基的六元杂环中, 这导致BPX和dtbpx电子效应的不同. 红外光谱和酸碱性研究证明, BPX的碱性比dtbpx弱很多, 因此向金属中心提供电子的能力降低.[(BPX)Pd(O2CCF3)2]晶体结构也证明六元杂环C-P-C 夹角(~106°)相比于dtbpx-Pd 配合物中C(tBu)-P-C(tBu)夹角(~110°)变小, 使得P原子孤对电子的σ-供体性质减弱, 而π-受体性质变强. 机理研究表明, 更缺电子的Pd-酰基物种有利于MeOH进攻和H+脱除, 从而加速醇解步骤的速率, 所以拥有亲电性更强的Pd中心的Pd-BPX表现出了高的催化活性, 而大体积BPX所创造的空间配位环境被认为是促进线性选择性的关键.
Fanjul等[91]基于dtbpx和dppx在乙烯羰化酯化反应中完全不同的活性和产物选择性, 设计了非对称结构的双齿膦配体A, 与Pd组成的乙烯羰化酯化催化剂具有优异的选择性和高活性, 并且比商用催化剂更稳定(图19). 通过对Pd、 Pt模型配合物的深入研究, 证明大体积的PtBu2基团控制着Pd-A配合物的立体结构, 其产生的重要结果是使关键中间体A1 的丙酰基处于PtBu2的反位(图20), 这和仅生成丙酸甲酯(MeP)的Pd-dtbpx体系的关键中间体类似.
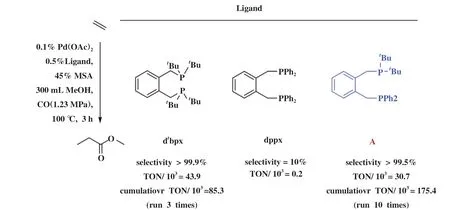
图19 乙烯羰化酯化不同Pd-P催化体系对照[91]Fig.19 Comparison of various Pd-P catalysts in the methoxycarbonylation of ethene[91]

图20 非对称双齿膦配体及化学选择性确定步骤中关键中间体结构[77]Fig.20 Unsymmetrical diphos ligands and the key intermediates[77]
在此基础上, Fanjul等[77,92]又相继发展了一系列基于邻二苯基骨架的非对称双齿膦配体(图20),并获得了一些初步的结构-活性关系, 也证明了对于非对称双齿膦-Pd配合物, 关键中间体A1相对于A2 的动力学优势是 MeP 出乎意料的高选择性的原因. Fanjul等的工作也说明邻二甲苯基双膦只需要一个体积庞大的P配位基即可形成非常有效的羰化酯化催化剂, 从而为配体设计提供了一些新思路.
2015 年, Holzapfel 和Bredenkamp[93]针对1-辛烯的羰化酯化反应比较了一系列膦配体的作用, 发现二茂铁骨架的非对称双齿膦配体表现出极好的催化活性和稳定性. 作者认为, 此类配体包含一个大体积缺电子P配位基和一个大体积富电子P配位基,并具有半稳定三齿配位的能力, 显示出反应速率和稳定性方面的优势.
3.3 钯-半稳定双齿膦配体催化体系
半稳定配体(hemilabile ligands)通常定义一类多齿配体, 其配位原子的给电子能力和极化度(软硬度)不同, 其中软配位原子形成较强配位键, 在反应中不易断开, 而硬配位原子配位键较弱, 易脱配位被取代. 常见的硬配位原子包括O、 N等杂原子,容易在催化循环中形成弱配位, 从而产生意料外的结果.
2017年, Beller课题组[94]在经典配体dtbpx(L13)和Ph2P(2-Py)(L2)的基础上开发了新型配体Pytbpx(L20), 希望能实现四甲基乙烯的选择性异构/羰化酯化反应. 根据Pd-H循环机理, 加入碱可促进醇解步骤, 但是不利于内烯异构, 因此在dtbpx配体中部分地引入两性基团(吡啶基)作为“质子梭子”来传递质子促进亲核试剂对钯-酰基中间体亲核进攻,加快醇解步骤的速率, 同时保留大体积叔丁基协同促进双键异构(图21 a). Pd-Pytbpx体系解决了多取代内烯的羰化酯化反应活性低的问题, 并且还能高效催化乙烯的羰化酯化反应, 反应TON > 1 425 000,TOF: 44 000 h-1, 总产率为85%, 选择性 > 99%.

图21 半稳定双齿膦-钯体系催化烯烃羰化酯化[94]Fig.21 Alkoxycarbonylation catalyzed by hemilabile bidentate P-Pd catalyst[94]
同年, Beller小组[95]又发展了一系列新型的二茂铁类半稳定双齿膦配体, 二茂铁骨架稳定、 可塑性强, 在P原子上连接空间位阻较大的两性基团, 提高了羰化酯化反应的活性, 使乙烯在80 ℃, 10 min内完全转化为丙酸甲酯. Pd-L21体系与Pd-dtbpx相比活化能大幅降低, 甚至在室温下也能实现乙烯和丙烯高效转为相应的酯, 为烯烃羰化酯化反应的工业应用开辟出新的催化体系(图21 b).
随后, Dong等[40]对该体系进行了机理研究, 并进一步研究了配体L21对羰化酯化反应实质性的影响. 作者提出一种类似于dtbpx-Pd体系的 Pd-H机制,所不同的是L21中2-吡啶基团具有多种功能. 一方面, 2-吡啶基作为“质子梭子”用于形成Pd-H物种和决速步骤的N辅助醇解, 形成稳定化的过渡态(图22, TS); 另一方面, 能够通过吡啶N原子与Pd中心的半稳定配位来提高催化剂的长期稳定性(图22,S-2D). 作者通过实验和DFT计算证实了这种金属-配体的协同作用.

图22 Pd-L21催化烯烃羰化酯化反应机理[40]Fig.22 Proposed mechanism for the alkoxycarbonylation using Pd-L21[40]
2019年, Beller小组[96]将非对称配体设计理念用于构建非对称的半稳定双齿膦配体HeMaRaphos(图23), 配体结构中大位阻、 富电子二叔丁基膦基团促进双键的快速异构化来实现高选择性, 叔丁基-2-吡啶基膦基团则促进钯氢活性物种的形成并加速醇解步骤实现高反应活性. HeMaRaphos与Pd配位实现了1,3-丁二烯的高选择性双羰化酯化直接制备己二酸二酯. 在工业上可行的条件下, Pd(TFA)2/HeMaRaphos/PTSA催化体系具有高达97%的选择性和100%的原子经济性, 转化数(TON)超过60 000, 并且能够实现200 g规模的己二酸二丁酯合成.
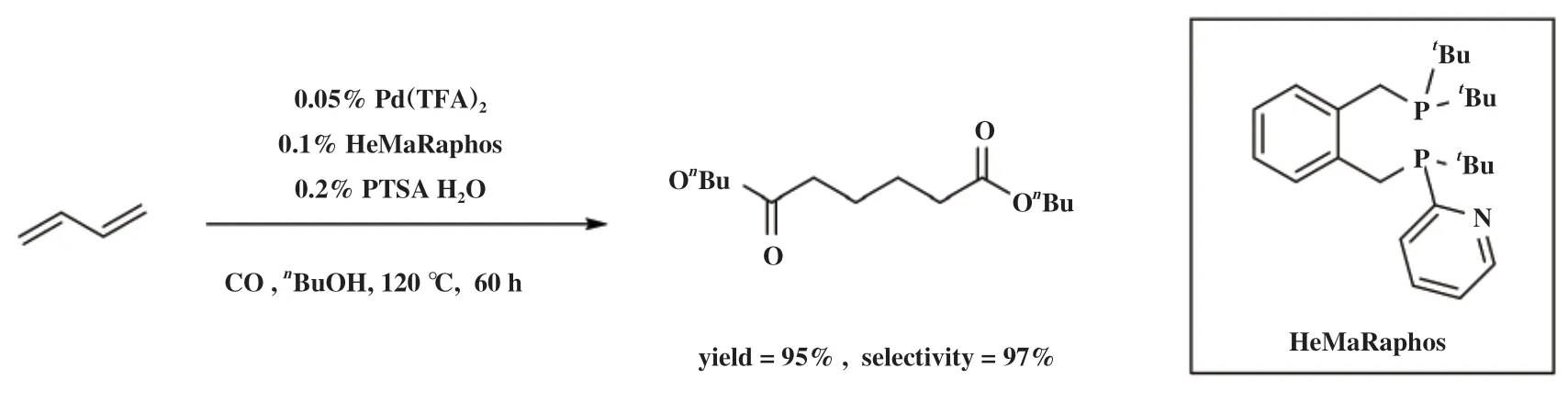
图23 Pd(TFA)2/HeMaRaphos/PTSA体系催化1,3-丁二烯双羰化酯化[96]Fig.23 Pd(TFA)2/HeMaRaphos/PTSA-catalyzed dicarbonylation of 1,3-dienes[96]
4 结论与展望
同时实现化学选择性和区域选择性是羰化酯化的一个挑战, 利用膦配体的电子效应和空间效应来影响和调控钯催化剂的活性和选择性是解决该问题的最有效途径. 在过去的几十年里, 围绕有机膦配体修饰的钯催化过程开展了大量的研究工作, 取得了一系列重要成果和进展. 例如, 膦配体的定性和定量配体参数成为研究配体效应的有力工具; 基于对配体效应、 催化机理深刻理解而发展的非对称双齿膦配体、 半稳定膦配体在羰化酯化反应中表现出卓越的性能, 具备了工业催化剂开发的条件.
虽然膦配体电子和空间效应对活性和选择性调控的研究成为新配体理性设计、 新催化体系构建的有利工具, 但是考虑到羰化酯化过程所涉及的复杂动力学, 反应速率取决于压力、 温度、 反离子和膦配体, 仍然很难预测实际的效果, 因此该领域尚有很多方面值得探索.
4.1 虽然Pd-H循环是普遍接受的机理, 但是,配体在关键步骤所起的作用仍然并不十分清楚, 仍需要开发更普适性的配体定性和定量配体参数, 从中寻找规律, 指导新配体设计.
4.2 钯催化羰化酯化过程和速率受到配体、 阴离子、 酸、 溶剂、 压力等因素影响, 对于一些特定底物, 如高级烯烃、 二烯烃, 配体的调控作用可能非常不同, 这些研究或将产生意料外的结果.
4.3 半稳定配位膦配体可直接参与底物或中间体的活化和/或识别, 与金属中心协同作用, 降低关键过渡态的能量, 加速速控步速率. 虽然内置的碱性基团被认为是增强活性的原因, 但直到现在它的确切作用还不十分清楚. 显然, 更好地理解这种实质性影响对于合理设计先进的工业催化剂具有重要意义.
4.4 随着理论计算方法的发展, 其在催化剂开发中越来越重要, 借助对已有催化反应的理解, 以及计算科学的进步, 加快发展膦配体计算方法和描述符, 建立配体库, 将为快速发现更优异的配体类型提供可能.
猜你喜欢
数字制造科学(2021年3期)2021-09-27 01:40:06
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
制造业自动化(2019年7期)2019-07-26 09:25:38
World Journal of Diabetes(2019年7期)2019-07-23 11:52:08
当代陕西(2019年6期)2019-04-17 05:04:10
纺织科学研究(2017年6期)2017-07-03 12:14:14
露天采矿技术(2017年5期)2017-06-05 15:06:16
纺织科学研究(2017年1期)2017-05-17 03:59:17
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
化工进展(2015年3期)2015-11-11 09:06:06
