
HPSEC法检测美罗培南中聚合物杂质的测定研究
2022-05-11 15:17:18陈丹丹戴红娟
江苏科技信息 2022年9期
孙 青,陈丹丹,戴红娟
(1.江苏省泰州医药高新技术产业园区管理委员会,江苏 泰州 225300;2.江苏农牧科技职业学院,江苏 泰州 225300;3.安若维他药业泰州有限公司,江苏 泰州 225300)
0 引言
美罗培南是广谱非肠给药的半合成碳青霉烯类抗生素,因其不仅对革兰阳性和革兰阴性菌均有较强的抗菌活性,而且对超广谱β内酰胺酶稳定,在临床多用于混合感染和严重感染的治疗。美罗培南最常见的不良反应是过敏反应,偶见过敏性休克;还可引起消化系统和皮肤系统等不良反应[1]。
美罗培南聚合物杂质产生的影响尚无研究,现行版中国药典也并未收载美罗培南聚合物杂质检测的方法[2]。因此,有必要建立有效检测美罗培南中聚合物杂质的方法,考察聚合物杂质产生的条件及稳定性,以控制聚合物杂质产生,有效降低不良反应。
近年来,高效分子排阻色谱技术逐渐成熟[3-4]。本文使用TSK gel G2000SWxL凝胶色谱柱,建立高效分子排阻色谱系统,测定美罗培南(见图1)中的聚合物杂质,并考察聚合物杂质的稳定性。
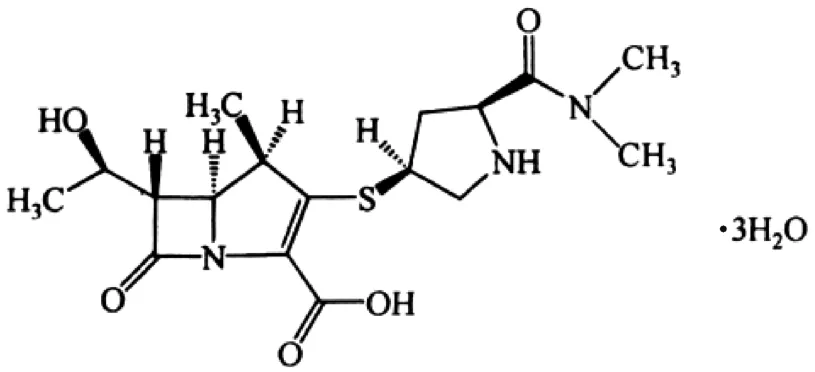
图1 美罗培南结构式
1 实验仪器与试药
实验仪器:Waters 2695高效液相色谱仪(沃特世公司);2998 PDA 光电二极管矩阵检测器(沃特世公司);Seven Easy pH 仪(Mettler公司);Mettler XS1O5DU型电子天平(Mettler公司)。
试药:美罗培南对照品(由中国食品药品检定研究院提供,产品批号130506—202004,含量以美罗培南 C17H25N3O5S 计86.8%);水为去离子水,乙腈为色谱纯,三乙胺、磷酸二氢钠、磷酸氢二钠、乙酸铵均为分析纯。
注射用美罗培南,山东罗欣药业集团有限公司,批号321033003,规格0.5 g。
2 方法与结果
2.1 色谱条件
色谱柱为TSK gel G2000SWXL凝胶色谱柱(7.8 mm×30.0 cm,5 μm),流动相0.1%三乙胺溶液(用磷酸调至中性)-乙腈(95∶5);流速0.8 mL/min;检测波长220 nm;柱温25 ℃;进样体积20 μL。
2.2 溶液的配制
精密称取美罗培南对照品适量,加水溶解并稀释制成每1 mL中约含美罗培南1 mg的溶液,作为美罗培南对照品溶液,备用。
精密称取注射用美罗培南样品适量,加水溶解并稀释制成每1 mL中约含美罗培南1 mg的溶液,作为供试品溶液,备用。
2.3 检测波长的选择
取2.2的溶液,采用Waters 2998二极管阵列检测器对上述1.0 mg/mL的对照品溶液进行全波长扫描,结果显示,在220和280 nm波长处主成分吸收较好。对比这两个波长发现,在220 nm处聚合物杂质峰吸收值更高,所以采用220 nm作为检测波长。
2.4 专属性试验
2.4.1 空白及辅料
为排除溶剂对检测的干扰,按2.1色谱条件进行液相分析,空白溶剂色谱图基线平稳,不干扰美罗培南的聚合物杂质测定。2.4.2 加速破坏试验
取2.2中的溶液,分别进行强光破坏(强光照射试验24 h、照度为5 000 lx)、紫外线破坏(254 nm紫外照射3 h)、高温破坏(60 ℃加热1 h)、氧化破坏(加入0.3%过氧化氢溶液1 mL,室温放置1 min)、酸破坏(加入0.1 mol/L盐酸溶液1 mL,室温放置1 min,用0.1 mol/L氢氧化钠溶液中和)、碱破坏(加入0.1 mol/L氢氧化钠溶液0.5 mL,室温放置1 min,用0.1 mol/L盐酸溶液中和),另一份不做任何破坏处理,作为对照,以上溶液按2.1色谱条件进行液相分析,记录色谱图(见图2)。结果显示,经过6种剧烈条件的破坏处理后,对照品中聚合物杂质含量均有不同程度的增加,几种破坏条件均可使聚合物杂质含量增加。聚合物杂质含量由高到低依次为高温破坏、酸破坏、紫外破坏、碱破坏、氧化破坏、强光破坏,经破坏处理后的聚合物杂质峰均能与美罗培南达到完全分离,表明该方法专属性良好。
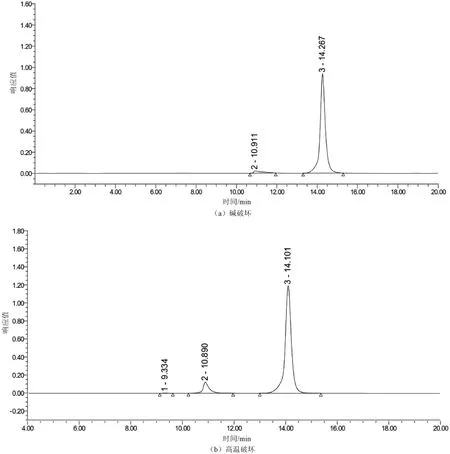
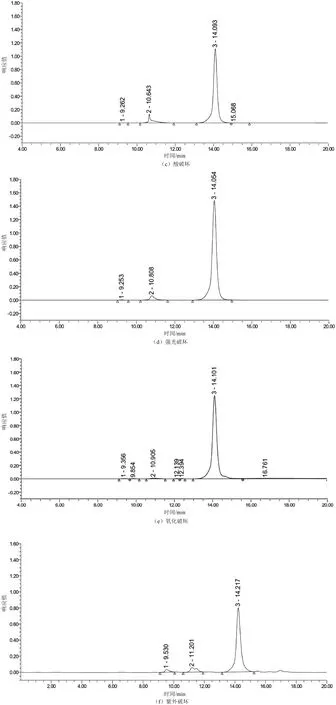
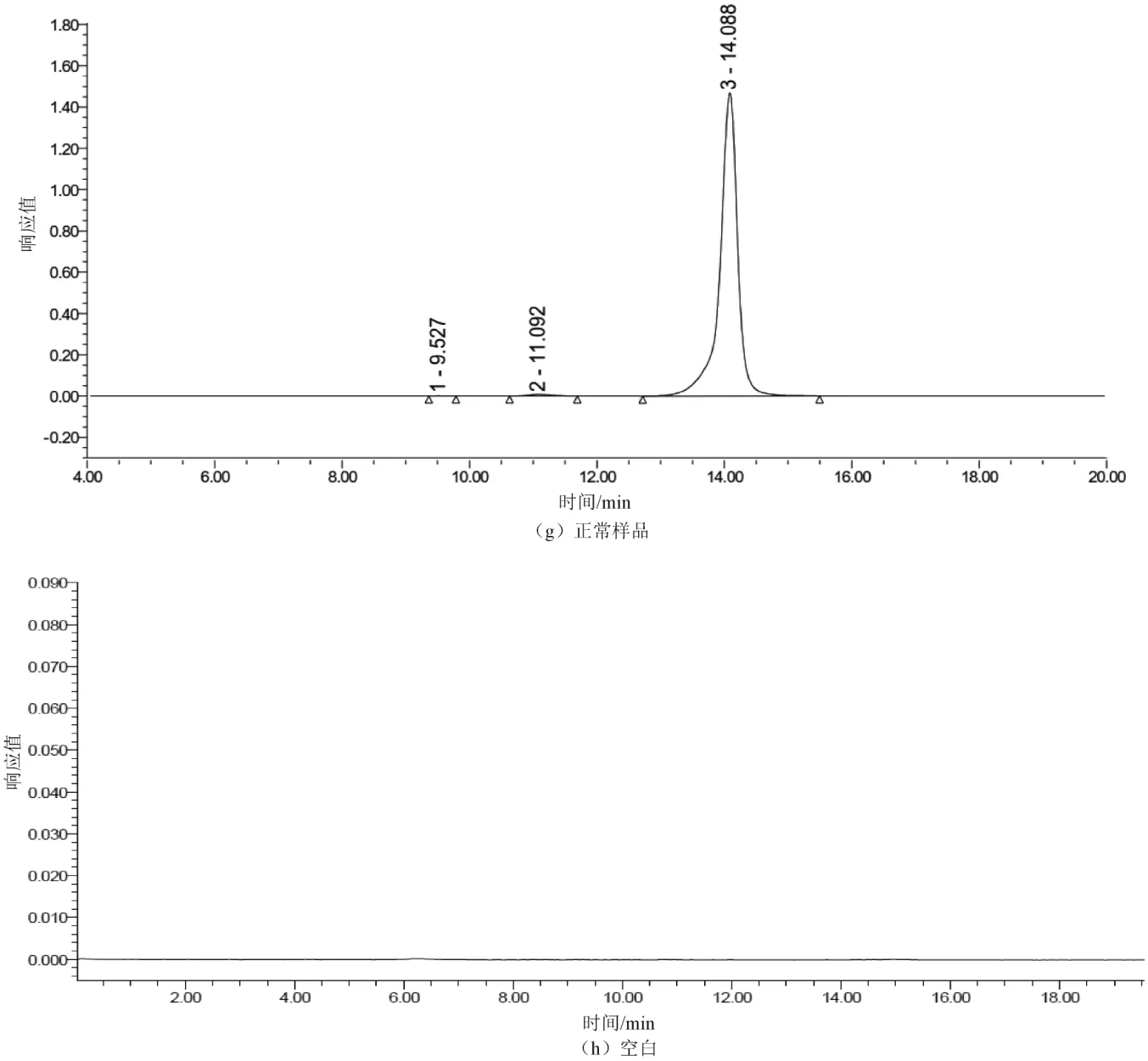
图2 加速破坏试验色谱图
2.5 系统适用性试验
参考专属性实验结果,在高温破坏条件下,有利于美罗培南聚合物杂质的产生,同时对主峰检测的影响较小,故本文采用高温破坏(60 ℃加热1 h)方法制备实验的系统适用性溶液。取2.2项下的溶液适量,60 ℃加热1 h,按照2.1色谱条件进行液相分析,记录色谱图。主成分美罗培南的保留时间为14.101 min,理论板数以美罗培南计为19 944,聚合物杂质峰1与聚合物杂质峰2的分离度为4.2,聚合物杂质峰2与美罗培南峰的分离度为7.7,均大于2.0,符合HPSEC系统适用性试验的分离度要求。
2.6 美罗培南峰线性考察
精密称取美罗培南对照品按美罗培南计,适量加水溶解并逐步稀释制成约含美罗培南1 976 μg/mL,988.0 μg/mL,494.0 μg/mL,197.6 μg/mL,98.80 μg/mL,49.40 μg/mL,19.76 μg/mL,9.880 μg/mL,1.976 μg/mL,0.988 μg/mL,0.494 μg/mL的对照品溶液。按照2.1色谱条件进行液相分析,以美罗培南溶液的浓度c为横坐标、 聚合物杂质的峰面积A为纵坐标绘制曲线,当美罗培南溶液质量浓度为0.000 5~2 mg/mL时,得到线性方程y=24 139 387x+52 881,相关系数R是
0.999 99,线性关系良好。
2.7 聚合物杂质峰线性考察
溶液配制同2.2,按2.1色谱条件进样,根据结果绘制曲线,横坐标为美罗培南的浓度,纵坐标为聚合物杂质峰面积总和。绘制散点图后进行线性曲线拟合,得到线性方程为y=157 009x-2 237.5(R=0.998),表明美罗培南溶液质量浓度为0.05~2 mg/mL时与聚合物杂质峰面积之和线性关系良好。
2.8 定量限与检测限
取2.7中的美罗培南对照品溶液,逐步稀释,按照2.1色谱条件进行液相分析,结果显示,测得的美罗培南的检测限(S/N=3)为0.49 μg/mL,定量限(S/N=10)为1.47 μg/mL,灵敏度良好。
2.9 进样精密度试验
取2.2项下对照品溶液,按照2.1色谱条件进行液相分析,连续进10针,测定美罗培南峰面积,对照品中美罗培南峰面积的相对标准偏差(RSD)为0.4%,进样精密度良好。
2.10 耐用性考察
2.10.1 流速的影响
分别设置流速为0.6 mL/min,0.8 mL/min,1.0 mL/min,按2.1色谱条件分析2.3中的系统适用性溶液,结果显示,在0.6~1.0 mL/min流速条件下,聚合物杂质峰均能与美罗培南峰有效分离,理论塔板数良好。考虑主成分峰的保留时间,最终选择流速为0.8 mL/min。
2.10.2 柱温的影响
分别设置柱温为20 ℃,25 ℃,30 ℃,结果柱温在(25±5)℃范围内均检测出两个聚合物杂质峰,且分离效果较好,方法耐用性较好。2.10.3 对照品稳定性考察
取新鲜配制的对照品溶液,在室温(约24 ℃)下放置24 h,按照2.1色谱条件进行液相分析,选取0 h,1 h,2 h,4 h,6 h,8 h,10 h,12 h,24 h时进样分析。结果表明:美罗培南主成分在室温下放置一段时间,峰面积变化不明显,RSD为1.2%;但美罗培南聚合物杂质峰面积增加显著,室温放置24 h内,聚合物杂质峰面积逐渐增加,24 h后为0 h的14倍。由结果可见,美罗培南水溶液不稳定,极易产生聚合物杂质,为了保证检测的可靠性,避免操作导致的误差,溶液必须现配现用。
2.11 样品聚合物的测定
取注射用美罗培南(山东罗欣药业集团有限公司,批号321033003),按照2.1色谱条件进行液相分析,记录色谱图。采用美罗培南自身对照外标法计算聚合物峰含量,美罗培南前的峰均为聚合物峰,该批注射用美罗培南聚合物含量为0.33%,参照2015年版中国药典,该值符合一般注射用β-内酰胺类抗生素的聚合物限度要求。
3 讨论
3.1 测定方法的选择
药典中记载的检测β-内酰胺类抗生素中高分子杂质的方法多为以葡聚糖G-10为填料的凝胶色谱法,该色谱柱存在柱效低、主峰和高分子杂质峰不能有效分离、分析时间较长,且不能使用有机溶剂等缺点[3-5]。本文建立以TSK gel G2000SWXL为色谱柱的高效分子排阻色谱法对美罗培南中的聚合物杂质进行分析,TSK gel G2000SWXL凝胶色谱柱填料主要为球状亲水改性硅胶,是一种新型分子排阻色谱柱,可分离相对分子质量500~15 000的高分子杂质,聚合物杂质主要在主峰前洗脱[4]。
由于聚合物杂质对照品难以获得,参考中国药典2020年版头孢米诺钠有关物质Ⅱ的检测,结合本实验对美罗培南主峰线性考察结果,本实验采用的是自身对照外标法来计算聚合物杂质含量。因注射用美罗培南样品较难获得,本文没有对更多厂家的样品进行聚合物含量的测定。
3.2 流动相的选择
在预实验中,作者分别以0.1%三乙胺溶液(用磷酸调至中性)-乙腈(95∶5)、磷酸盐缓冲液(pH 7.0) -乙腈(95∶5)、0.01 mol/L乙酸铵溶液(冰醋酸调至中性)-乙腈(95∶5)为流动相进行液相分析比较。结果发现,以0.1%三乙胺溶液-乙腈(95∶5)为流动相时,美罗培南主峰前可分离出两个杂质峰,主峰与各高分子杂质峰的分离度最好,故本文采用0.1%三乙胺溶液(用磷酸调至中性)-乙腈(95∶5)为流动相。
3.3 美罗培南稳定性的考察
本文建立一种有效的色谱系统测定美罗培南中的聚合物杂质含量,并进行方法学验证。由2.5可知,美罗培南易产生聚合物杂质,在254 nm紫外光照射24 h后,美罗培南主成分峰几乎全部被破坏,产生大量的聚合物杂质,因此美罗培南在储存、运输及使用过程中应尽量避免紫外光的照射,除紫外光外,美罗培南在碱性、高温及酸性条件下也容易产生聚合物杂质。由2.10.3可知,美罗培南的水溶液在室温条件下极易产生聚合物杂质,放置24 h后,聚合物杂质峰面积增长约14倍,因此,注射用美罗培南生产过程中应严格控制配液、除菌过滤、制剂灌装的工序时间。医疗机构使用美罗培南时应现配现用,从而减少美罗培南聚合物杂质的产生,降低不良反应发生率,提升药品质量,保障人民群众用药安全。
猜你喜欢
中西医结合心脑血管病杂志(2022年12期)2022-07-07 10:24:24
中国典型病例大全(2022年7期)2022-04-22 00:50:34
艺术品鉴(2020年6期)2020-12-06 10:49:08
中华养生保健(2020年2期)2020-11-16 00:49:20
领导文萃(2017年6期)2017-03-24 09:31:39
中学生数理化·高一版(2016年7期)2016-12-07 20:47:07
中国卫生标准管理(2015年3期)2016-01-15 02:49:19
中学生数理化·中考版(2015年12期)2015-09-10 07:22:44
云南中医学院学报(2015年1期)2015-07-31 18:10:45
中国药业(2014年12期)2014-06-06 02:17:13
