
LC-MS/MS法测定人血浆中普芦卡必利的浓度
2022-05-11 07:07李长印廖健城陆明霞宋慧婷居文政邹建东储继红
中国药理学通报 2022年5期
李长印,廖健城,陆明霞,宋慧婷,居文政,邹建东,储继红
(南京中医药大学附属医院,江苏省中医院,临床药理科,江苏 南京 210029)
琥珀酸普芦卡必利(prucalopride succinate)为苯并呋喃类促肠动力药,是特异性的5-HT4受体完全激动剂,对5- HT4受体具有较高的选择性。它可通过增加胆碱能神经递质的释放,刺激肠蠕动反射,增强结肠收缩和近端结肠传输,从而有效缓解便秘病人的症状[1-2]。琥珀酸普芦卡必利片由比利时Movetis公司研制,CFDA于2012年12月31日批准了该药的进口注册申请,片剂,规格为1 mg、2 mg,商品名为力洛(Resolor),用于治疗成年女性患者中应用轻泻剂难以充分缓解的慢性便秘症状。
前期研究表明,琥珀酸普芦卡必利片进入人体后主要以普芦卡必利(prucalopride,PCP)的形式被检测到[3-7]。近年来,虽然PCP的人体药代动力学和生物等效性的相关研究已有一些报道[4-6],对研究中所采用的测定方法如LC-MS/MS法[4, 6]和放射免疫法[5]也有提及,但目前尚无完整系统的人血浆中PCP浓度测定的方法学研究报道。LC-MS/MS法是目前检测生物样品中药物浓度的最常用测定方法[4, 6, 8-9],Zuo等[3]在PCP大鼠药代动力学和组织分布的一项研究中考察了大鼠血浆PCP浓度的LC-MS/MS测定方法学,但其中缺乏对溶血、高脂基质效应和全血稳定性等考察项目的验证。同时,大鼠血样和人血样的基质也存在着诸多差异,有可能对PCP的测定产生影响。基于此,本研究旨在建立并验证一种可靠的PCP人血药浓度LC-MS/MS测定方法,以满足PCP人体药代动力学和生物等效性研究中的检测需求,为相关药物制剂的研发提供方法学支持。
1 材料
1.1 药品与试剂琥珀酸普芦卡必利对照品(济川药业集团有限公司,批号160328,纯度99.9%,分子式为C18H26ClN3O3·C4H6O4,结构式见Fig 1A);PCP-13CD3(dPCP)对照品(内标,美国Stanta Cruz公司,批号E2517,纯度99.7%,同位素丰度99.5%,分子式为13CC17H23D3ClN3O3,结构式见Fig 1B);乙酸铵(ACS公司,HPLC级,批号50Y1408GV);甲醇(Merck Company,HPLC级);超纯水由Millipore Milli-Q Advantage A10超纯水机制备;乙酸乙酯(南京化学试剂股份有限公司,分析纯,批号160927513F);氢氧化钠(南京化学试剂股份有限公司,分析纯,批号11021820130);琥珀酸普芦卡必利片(商品名:力洛®,Jassen Clig S.p.A公司,规格2 mg/片);人空白血浆(江苏省血液中心);人空白全血(江苏省中医院);甘油三酯(Sigma-Aldrich,批号SLBZ5252,纯度≥99%);甘油(国药集团化学试剂有限公司,分析纯,批号20191226)。
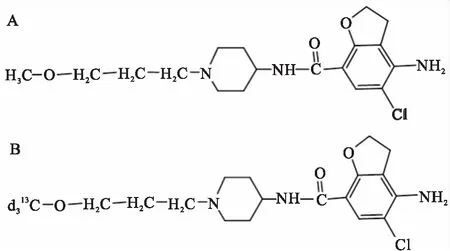
Fig 1 Chemical structures of prucalopride (A) and prucalopride-13CD3 (B)
1.2 实验仪器液相色谱质联用仪,包含Pump Agilent 1260 G1312A,Column Oven Agilent 1260 G1316A,Auto Sampler Agilent 1260 G1367E和AB Sciex Mass spectrometer API4000,液质分析检测系统由软件AB Sciex Analyst 1.6控制;十万分之一电子天平(美国Mettler Toledo公司,型号MS105);万分之一电子天平(上海菁华公司,型号FA2004N);常速离心机(常州国华电器有限公司,型号80-4);高速离心机(德国Eppendorf公司,型号5417R;美国Thermo公司,型号Micro 17R);微型旋涡混合仪(上海沪西分析仪器厂有限公司,型号WH3);离心浓缩仪(美国Labconco公司,型号CentriVap);-80 ℃超低温保存箱、-20 ℃冷藏冷冻转换柜、4 ℃医用冷藏箱均为青岛海尔特种电器有限公司生产;移液器(德国Eppendorf公司,规格20 μL/100 μL/200 μL/1000 μL/5000 μL)。
2 方法与结果
2.1 液相色谱与质谱条件液相色谱条件:Agilent ZORBAX SB-C18色谱柱(3.0×100 mm,3.5 μm),Agilent ZORAX SB-C18保护柱(4.6×12.5 mm,5 μm),采用等度洗脱,流动相为含1 mmol·L-1乙酸铵的水:甲醇 (20 ∶80,V/V),流速400 μL·min-1,柱温35 ℃,分析时间5 min,进样体积4 μL。
质谱检测条件:电喷雾离子源,正离子模式下采用多反应监测(MRM)模式采集数据,PCP的检测目标离子对为m/z368.4/196.0,去簇电压(DP)为96 V,碰撞能量(CE)为40 eV,dPCP的检测目标离子对为m/z374.4/198.0,DP为91 V,CE为43 eV。Dwell Time 200 ms,碰撞室入口电压(EP)10 V,碰撞室出口电压(CXP)14 V,碰撞气(CAD)10 psi,气帘气(CUR)25 psi,辅助气1(GS1)60 psi,辅助气2(GS2)65 psi,电喷雾电压(ISV)5 500 V,源温(TEM)500 ℃。
2.2 标准溶液的配制
2.2.1PCP对照品储备液及工作液的配制 分别精密称取琥珀酸PCP对照品10.69 mg和10.41 mg,经质量校正后分别相当于含PCP 8.068 mg和7.856 mg。分别以甲醇溶解、定容至10 mL,摇匀即得浓度为0.806 8 g·L-1的PCP标准曲线储备液I和浓度为0.785 6 g·L-1的PCP质控储备液II,将两者储存于4 ℃冰箱中备用。
用甲醇将PCP储备液I进行稀释,获得浓度依次为1.179、2.358、4.717、9.434、18.87、37.74、75.47、150.9 μg·L-1的PCP系列标准曲线工作液;用甲醇将储备液II进行稀释,获得浓度依次为1.179、3.275、14.74、117.9 mg·L-1的PCP系列质控工作液。所有工作液4 ℃冰箱中储存备用。
2.2.2内标dPCP储备液及工作液的配制 取由Santa Cruz公司精密称量的dPCP对照品1瓶(1.000 mg),精密加入1 000 μL甲醇溶解,摇匀,即得1.000 g·L-1的dPCP储备液;将此储备液稀释100 000倍,即得浓度为10 μg·L-1的dPCP工作液。两者均冷藏于4 ℃冰箱中备用。
2.3 血浆样品处理
2.3.1空白血样及受试者含药血浆样品的处理 精密吸取空白血浆(或受试者含药血浆)400 μL于10 mL玻璃管中,依次加入甲醇(或10 μg·L-1的dPCP工作液)100 μL,1 mol·L-1氢氧化钠溶液80 μL,乙酸乙酯4 mL,涡旋10 min进行充分提取,2 000 r·min×10 min离心,吸取上清液3 mL,于离心浓缩仪中40 ℃挥干,加入80%甲醇水200 μL,涡旋1 min混匀,12 000g×5 min,4 ℃离心,取上清液4 μL进行LC-MS/MS分析。
2.3.2标准曲线血浆样品的配制与处理 取空白血浆400 μL于10 mL玻璃管中,分别依次精密加入系列标准曲线工作液20 μL,涡旋混匀,即得浓度依次为0.058 96、0.117 9、0.235 8、0.471 7、0.943 4、1.887、3.774、7.547 μg·L-1的系列PCP标准曲线血浆样品。后续处理同“2.3.1”项。
2.3.3质控血样的配制与处理 分别取空白血浆400 μL于10 mL玻璃管中,分别精密加入系列质控工作液20 μL,涡旋混匀,即得浓度依次为0.058 96、0.163 8、0.737、5.896 μg·L-1的系列PCP质控血样。后续处理同“2.3.1”项。
2.4 分析方法的验证
2.4.1方法的选择性 如Fig 2A所示,在该方法设定的LC-MS/MS分离检测条件下,溶液及血浆样品中PCP和dPCP的色谱峰形良好,出峰时间稳定在3.6 min左右;如Fig 2B所示,不含内标的定量上限样本中待测物在内标检测离子通道无明显干扰;如Fig 2C所示,工作液浓度的内标dPCP在待测物PCP检测离子通道无明显干扰;如Fig 2D-F所示,空白血浆、含药血浆及待测血浆样品中PCP和dPCP检测离子通道均无明显干扰峰出现。上述考察结果表明,该方法特异性良好,血浆基质中的内源性化合物和代谢物不影响PCP的准确定量,待测物和内标间无影响检测的相互干扰。此外,在标准曲线的定量上限样品之后进样分析的双空白血浆样品中未见明显的PCP和dPCP干扰峰,表明该方法无明显系统残留。

Fig 2 Typical LC-MS/MS chromatograms of prucalopride (PCP) and prucalopride-13CD3 (dPCP)
2.4.2基质效应 按“2.3.1”项处理6份不同来源的正常空白血浆、1份溶血基质空白血浆(由空白血浆加入4%空白全血混合配制而成)、1份高脂基质空白血浆(称取甘油三酯30.63 mg溶解在0.2 mL甘油中,再加入空白血浆9.80 mL,混匀即得),获得其相应的吹干样品,用于配制未经提取的含药基质样品。分别向其中加入20 μL的低、高浓度PCP质控工作液,再依次加入dPCP工作液100 μL、50%甲醇水溶液80 μL,涡旋1 min混匀,12 000g×5 min,4 ℃离心,吸取上清液4 μL进行LC-MS/MS 分析。每个浓度平行制备6个样品。记录色谱图、PCP色谱峰面积As和dPCP色谱峰面积Ai。

2.4.3标准曲线与线性范围 按照“2.3.2 ”项制备并检测标准曲线血浆样品,记录色谱图、PCP色谱峰面积As和dPCP色谱峰面积Ai。以PCP血药浓度C为X轴,以As与Ai的比值f(f=As/Ai)为Y轴,以1/C2作为权重系数,进行线性回归。最终标线方程为y=(1.22±0.04)x+(0.0285±0.003 98)(n=5),相关系数r为0.996 3~0.999 6。由上述结果可知,在0.058 96~7.547 μg·L-1的血药浓度范围内,PCP色谱峰面积比值与其浓度的线性关系良好。
2.4.4精密度和准确度考察 按“2.3.3”项制备并检测PCP系列质控血样,每个浓度平行6个样品,连续制备并检测3批。记录色谱图、PCP色谱峰面积As和dPCP色谱峰面积Ai。计算峰面积比值f(f=As/Ai),进而将f值代入随行标线方程,求得各质控血样的实测浓度及其准确度。批内精密度以第一批质控血样(n=6)的实测浓度值的CV表示,批间精密度用所有3批质控血样(n=18)的实测浓度值的CV表示。Tab 1汇总了精密度和准确度考察的详细测定结果。结果表明,本方法的批内及批间准确度良好,且精密可重现。
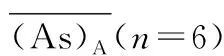
2.4.6稳定性考察
2.4.6.1 待测物储备液稳定性 精密吸取按“2.2”项新鲜配制的PCP储备液a和dPCP储备液、已于室温避光放置6 h后的PCP储备液b、已于4 ℃冰箱中放置59 d的储备液c适量,分别用甲醇将3种PCP储备液稀释至3.019 μg·L-1,将dPCP储备液稀释至10 μg·L-1,而后将3种PCP稀释液分别和dPCP稀释液等体积混合均匀后进样分析,每种PCP储备液平行配制6个样品。记录色谱图、化合物色谱峰面积。考察新鲜配制储备液与放置后PCP储备液的峰面积偏差和CV(n=6),峰面积偏差=(放置的储备液峰面积均值-新鲜配制的储备液峰面积均值)/新鲜配制的储备液峰面积均值。结果显示:与新鲜配制的PCP储备液相比,放置6 h和59 d后PCP储备液的峰面积偏差分别为3.11%和-5.33%,CV分别为1.93 %和2.51%。结果表明,待测物PCP储备液室温避光放置6 h及4 ℃冰箱中放置59 d均稳定。
2.4.6.2 待测物和内标工作液稳定性 精密吸取按“2.2”项新鲜配制的浓度分别为1.179和150.9 μg·L-1的PCP工作液各20 μL,依次加入新鲜配制的浓度为10 μg·L-1的dPCP工作液 100 μL及甲醇80 μL,混合均匀后,作为新鲜配制的工作液稳定性考察用样品,进行LC-MS/MS分析,平行配制6份。同法制备分析室温避光放置6 h的工作液稳定性样品,以及4 ℃冰箱中放置12 d的工作液稳定性样品。记录色谱图、化合物色谱峰面积。计算工作液放置后与新鲜配制时的色谱峰面积偏差及其CV(n=6),其中峰面积偏差=(放置后工作液峰面积均值-新鲜配制工作液峰面积均值)/新鲜配制工作液峰面积均值。测定结果显示:与新鲜配制时相比,浓度为1.179和150.9 μg·L-1的PCP工作液放置6 h后,其色谱峰面积偏差分别为0.56%和1.32%,CV分别为2.97%和1.48%;放置12 d后其峰面积偏差分别为2.36%和-1.10%,CV分别为4.35%和3.24%;浓度为10 μg·L-1的dPCP工作液放置6 h后的峰面积偏差为4.04%,CV为3.87%。上述考察结果表明,待测物PCP工作液室温避光放置6 h及4 ℃冰箱中放置12 d均稳定,内标dPCP工作液室温避光放置6 h稳定。
2.4.6.3 血浆样品稳定性 按“2.3.3”项制备低、高浓度PCP质控血样,分别考察室温放置6 h、-80 ℃反复冻融3次、-80 ℃冻存56 d、-20 ℃冻存60 h的血浆样本稳定性,以及处理后样品于自动进样器(8 ℃)中放置24 h的稳定性。每个存放条件下每个浓度平行6个样品。结果见Tab 2,表明血浆样品在上述各种存放条件下的稳定性良好。

Tab 1 Intra- and inter-batch precision and accuracy for determination of PCP in human plasma

2.4.6.4 全血样品稳定性 分别精密吸取浓度为3.275、117.9 μg·L-1的质控工作液50 μL于1 000 μL空白全血中,混匀即得浓度0.163 8、5.896 μg·L-1的低、高浓度全血样品。每个浓度平行6个样品为1组,共制备2组。1组立即离心,取上层血浆400 μL,按“2.3.3”项进行样品制备并检测;另一组室温放置4 h后离心,取上层血浆400 μL同法制备样品并检测。记录色谱图、PCP色谱峰面积As和dPCP色谱峰面积Ai,计算色谱峰面积比值f(f=As/Ai),将f值代入随行标线方程求得各样品实测浓度及其准确度,并计算CV(n=6)。测定结果显示:低浓度全血样品0 h和4 h实测浓度分别为(0.170 0±0.006 619)和(0.157 7±0.008 632)μg·L-1,准确度均值分别为103.8%和96.30%,CV分别为3.89%和5.47%;高浓度全血样品0 h和4 h实测浓度分别为(5.843±0.087 69)和(5.328±0.423 3)μg·L-1,准确度均值分别为99.10%和90.36%,CV分别为1.50%和7.95%。结果表明,PCP在全血样品基质中室温放置4 h稳定性良好。
2.5 应用女性健康受试者24名,经体检合格后,签署知情同意书,并通过南京中医药大学附属医院伦理委员会审批,进行人体药代动力学试验。各受试者口服琥珀酸普芦卡必利片1片,分别于给药前、给药后0.5、1、1.5、2、2.5、3、4、5、6、8、12、24、48和72 h经肘静脉取血4 mL置于柠檬酸钠化的采血管中,混和均匀,4 ℃离心,3 000 r·min-110 min,吸取上层血浆,按“2.3.1”项制备血样,并进行LC-MS/MS分析,检测受试者血样中的PCP药物浓度。浓度测定结果显示,服药后各受试者的PCP血药浓度在0.075 9~5.455 μg·L-1之间,均处于本方法的线性范围(0.058 96~7.547 μg·L-1)内。该试验结果表明,本研究所建立并验证的LC-MS/MS分析方法适用于检测人血浆中PCP药物浓度,可满足PCP人体药代动力学和生物等效性研究的需要。
3 讨论
在方法学建立和优化过程中,作者对不同的色谱柱类型、流动相组成、样品前处理方法和质谱检测条件等进行了多种尝试。结果表明:① Agilent ZORBAX SB C18色谱柱的色谱峰形和分离效果优于Alltima HP C18、Chrom-matrix innovationTMUltimate C18和Agilent Poroshell 120 SB-C18等色谱柱。② 在水相流动相中加入终浓度为1 mmol·L-1的乙酸铵,可使PCP及dPCP的保留时间稳定,具有良好的色谱峰峰形和响应;流动相中如果加入甲酸会导致保留时间明显前移,加入氨水会使其后移,加入甲酸铵则会使保留时间不稳定,三者间的各种组合及其与乙酸铵的组合均没有单用1 mmol·L-1乙酸铵好。③蛋白沉淀法不能满足当前仪器的灵敏度要求;乙酸乙酯提取回收率优于乙醚和二氯甲烷,提取过程中加入NaOH溶液可提高乙酸乙酯提取效率,最佳加入浓度和量分别为1 mol·L-1和80 μL。④由于PCP结构中含有氯元素(Fig 1),其同位素离子也具有较高的丰度,导致PCP可能对同位素内标产生干扰,因此作者选用内标dPCP的同位素离子[M+2+H]+作为内标监测离子对的母离子,从而避免了分析物对内标产生的干扰。
本研究首次建立了基于乙酸乙酯液液萃取的人血浆中PCP浓度的LC-MS/MS测定方法,并对该方法的选择性、基质效应(包括溶血和高脂)、线性、精密度和准确度、提取回收率、稳定性(包括全血稳定性)等进行了系统验证,结果表明:该方法简单、可靠、灵敏、稳定、耐用。与文献报道的PCP血浆浓度的LC-MS/MS测定方法相比,本方法的优势在于:① 与前期研究多采用蛋白沉淀法处理样品相比[3-4],本方法采用乙酸乙酯萃取所得进样样品更为“干净”,更适用于临床研究的高通量样品分析;同时借此可对待测物PCP进行一定程度的浓度富集,对仪器本身的灵敏度要求不高,使得方法更具有普遍适用性。② 通过对流动相、样品前处理方法等多环节的摸索优化,方法的灵敏度有所提高(从0.1 μg·L-1~0.058 96 μg·L-1)[3, 5]。③ 与前期研究不同,采用同位素内标dPCP有助于消除基质效应,使得该方法更耐用;同时采用内标的同位素离子作为监测离子可避免PCP对内标检测的干扰,使得该方法更可靠。上述优势保证该方法能够准确快速地测定人血浆中PCP药物浓度,适用于PCP相关制剂的人体药代动力学和生物等效性研究。
猜你喜欢
云南化工(2022年6期)2022-06-29
氯碱工业(2021年12期)2021-12-27
口腔护理用品工业(2021年4期)2021-11-02
东坡赤壁诗词(2021年1期)2021-03-24
幸福家庭(2020年8期)2020-07-27
当代临床医刊(2020年2期)2020-06-17
发明与创新·中学生(2019年9期)2019-09-12
伴侣(2019年11期)2019-08-09
中国科技纵横(2019年23期)2019-02-14
海峡科技与产业(2017年1期)2017-03-04
