
RIPK3介导坏死样凋亡参与心肌缺血/再灌注损伤研究进展
2022-05-11 07:07:20阴苏月王守宝杜冠华
中国药理学通报 2022年5期
阴苏月,王守宝,杜冠华
(中国医学科学院北京协和医学院药物研究所,药物靶点研究与新药筛选北京市重点实验室,北京 100050)
重建冠脉血流,缓解心肌血流灌注不足,降低耗氧量,以维持冠脉血流和心肌需求的相对平衡,是缺血性心脏病的主要治疗手段。心肌缺血/再灌注损伤(myocardial ischemia reperfusion injury,MIRI)是因阻塞的冠状动脉重新开放,出现的心肌细胞不可逆性死亡的加重和梗死区扩大,这在临床实践中也被称为“无复流”现象[1]。MIRI增加了缺血性心脏病患者心脏不良重构的发生率,也是该类患者远期存活率较低且预后较差的重要原因,但目前尚无有效的治疗方法[2]。
细胞凋亡和坏死被认为是参与MIRI的两条主要细胞死亡途径。近年来,研究人员发现了一种新的细胞死亡途径——坏死样凋亡,其既有与细胞凋亡类似的可调控途径,又有细胞发生坏死后的形态学特征。坏死样凋亡主要由受体相互作用蛋白激酶1(receptor interacting protein kinase1,RIPK1),RIPK3及RIPK3下游效应子混合谱系激酶结构域样蛋白(mixed-lineage kinase domain-like pseudokinase,MLKL)来介导,不依赖于caspase的细胞死亡形式[3]。
坏死样凋亡在细胞死亡途径研究中受到越来越多的关注。研究表明,RIPK1和RIPK3都可以通过凋亡和坏死样凋亡两种机制诱导细胞死亡。坏死样凋亡发生的两个基本条件是细胞表达RIPK3和caspase-8被抑制,而抑制caspase-8会导致RIPK3的激活。当RIPK3被敲除时,RIPK1只能引起细胞凋亡,若RIPK3存在,RIPK1则可以引起细胞发生坏死样凋亡,且RIPK3在下游效应蛋白MLKL大量存在时,可以单独介导坏死样凋亡的发生[4]。这些都表明了RIPK3对于坏死样凋亡中是必不可少的。RIPK3介导的坏死样凋亡参与了包括心脏在内的不同器官的多种病理情况,并能参与多种病理条件下的细胞死亡,如病毒感染、急性肾损伤和缺血/再灌注损伤[5]。
研究发现,RIPK3介导的坏死样凋亡在MIRI病理进程中发挥着重要作用,因此靶向RIPK3介导的坏死样凋亡可能成为一种新颖的、有效的干预策略。详细了解RIPK3介导的坏死样凋亡的发生机制及其参与MIRI发病的具体过程,将有助于研究在疾病的发生发展和后期治疗中,心肌细胞的相互作用,并将RIPK3介导坏死样凋亡的潜在益处转化为临床研究。
1 RIPK3的结构与生物功能
RIPK家族是一类具有丝氨酸/苏氨酸激酶活性的蛋白家族。其家族成员N端包含同源激酶结构域,并具有经典的催化元件,如同源但不相同的P-环(P-loops),位于αC螺旋中心的赖氨酸和谷氨酸离子对,以及催化环中的HXD基序[6]。但RIPK家族的C端却各不相同,可特异性地与蛋白结合发挥不同的生物功能。不同于RIPK1 C端的一段死亡结构域(death domain,DD),RIPK3的C端没有DD结构,但有一段和RIPK1同样的片段,即RIP同型结构域(RIP homotypic interaction motif,RHIM)。这个结构域奠定了RIPK3和RIPK1相互作用的基础,对坏死样凋亡的发生至关重要[6-7]。
RIPK3在调节细胞存活和死亡中发挥重要作用。最初认为RIPK3的作用是介导核因子-κB(NF-κB)的激活和细胞凋亡,但在后续研究中发现RIPK3在介导坏死样凋亡中起到不可或缺的作用[5]。结构完整的RIPK3参与了依赖caspase和不依赖caspase的细胞程序性死亡。当caspase-8在RIPK3的Asp328位点处剪切掉其特异结构域后,RIPK3只能介导依赖caspase发生的细胞凋亡,这说明结构完整RIPK3的激活在不依赖caspase的细胞死亡途径中是必不可少的[8]。在RIPK3-/-细胞中,RIPK1无法和RIPK3形成坏死复合体,继而不能引发坏死样凋亡[9]。这些都表明RIPK3在坏死样凋亡中的重要性。
RIPK3在炎症反应中也起着重要的作用,涉及多种疾病如:缺血/再灌注损伤、脓毒症、神经退行性疾病、胰腺炎、胃肠炎和皮炎等。有报道称RIPK3可以特异性激活依赖于NLRP3-caspase-1的炎症,而且RIPK3可以激活炎症小体[5, 10]。RIPK1缺乏引起的剧烈炎症反应会导致围产期小鼠死亡,但敲除RIPK3可以降低其死亡率[11]。而且越来越多研究发现,敲除RIPK3可以逆转炎症性疾病及相关损伤,故与RIPK1抑制剂相比,RIPK3抑制剂更有可能保护细胞使其免受来自各方面的刺激。
综上,RIPK3的主要功能是介导细胞坏死样凋亡,其依赖于RIPK3结构中具有激酶活性的结构域[7]。除此之外,RIPK3还有不依赖激酶活性结构域的功能,这些功能与炎症小体的激活和细胞凋亡有关[12]。因此,RIPK3在调节坏死样凋亡和炎症反应中有着不容忽视的作用。
2 RIPK3介导坏死样凋亡的途径
坏死样凋亡可被多种信号激活,如肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)、DNA损伤、脂多糖(lipopolysaccharide,LPS)、缺氧等[13]。调控坏死样凋亡的过程非常复杂且精密,目前研究较为清晰的有以下两种(Fig 1)。
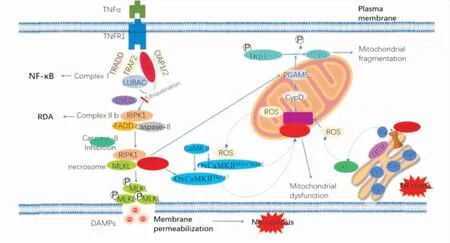
Fig 1 Necroptosis and RIPK3-related pathways in myocardial ischemia-reperfusion injury
2.1 RIPK3通过MLKL介导的坏死样凋亡通路坏死样凋亡可由多种受体启动,如:死亡受体肿瘤坏死因子受体1(tumor necrosis factor receptor 1,TNFR1),模式识别受体Toll样受体(Toll-like receptors,TLR)等[5, 14]。RIPK3对巨噬细胞坏死样凋亡的调节与不同受体的激活有关,其活化常常出现在TNF诱导的细胞通路中。
TNFR1受到TNF-α刺激后,首先招募肿瘤坏死因子受体相关死亡结构域(TNF receptor associated death domain,TRADD)、RIPK1和TNF受体相关因子2(TNF receptor-associated factor 2,TRAF2),而后TRAF2和细胞凋亡蛋白抑制因子1/2(cellular inhibitor of apoptosis protein 1 and 2,cIAP1/2)结合,共同形成复合体Ⅰ。RIPK1的泛素化主要由cIAP1/2和线性泛素链组装复合体(linear ubiquitin chain assembly complex,LUBAC)来介导,泛素化后的RIPK1可以激活NF-κB通路,启动细胞存活通路及炎症反应。当NF-κB信号通路被抑制时,复合体Ⅰ中的TRADD招募Fas相关死亡结构域蛋白(Fas-associated death domain protein,FADD)和caspase-8,形成复合体Ⅱa,此时发生不依赖RIPK1的凋亡(RIPK1 independent apoptosis,RIA);当去泛素化酶,如肿瘤抑制因子头帕肿瘤综合征蛋白(cylindromatisis,CYLD),使RIPK1去泛素化,RIPK1招募FADD和caspase-8后形成复合物Ⅱb,介导依赖RIPK1的凋亡(RIPK1 dependent apoptosis,RDA)。当启动细胞凋亡的caspase-8被抑制的时候,caspase-8对RIPK1的抑制作用也会随之解除,RIPK1和RIPK3通过RHIM相互作用,形成RIPK1-RIPK3复合体(necrosome),RIPK1磷酸化RIPK3,RIPK3进一步磷酸化MLKL,磷酸化的MLKL发生寡聚和移位,最终在质膜上执行坏死样凋亡[14]。
当病毒RNA、LPS和dsRNA激活TLR3或TLR4时,β干扰素TIR结构域衔接蛋白(TIR-domain containing adapter inducing interferon-β,TRIF)被特异性招募,通过其结构域中特有的RHIM形成RIPK3/TRIF复合物,在caspase-8抑制剂或泛caspase抑制剂(zVAD-FMK)存在的情况下,形成坏死小体,继而细胞发生坏死样凋亡[15]。
2.2 RIPK3通过CaMKII介导的坏死样凋亡通路RIPK3除了通过上述RIPK1-RIPK3-MLKL级联反应引发坏死样凋亡外,还可通过激活钙/钙调蛋白依赖性蛋白激酶Ⅱ(calcium/calmodulin-dependent protein kinaseⅡ,CaMKII)诱导细胞发生坏死样凋亡[16]。CaMKII是一种主要的钙调节蛋白,参与MIRI、心律失常和心力衰竭等疾病发生发展的过程[16]。RIPK3可直接磷酸化CaMKII的第287位苏氨酸,而RIPK3通过内质网(endoplasmic reticulum,ER)-Ca2+-黄嘌呤氧化酶(xanthine oxidase,XO)信号通路诱导并过量产生的细胞内活性氧(reactive oxygen species,ROS),也可使CaMKII的第281/282位蛋氨酸发生氧化,激活的CaMKII可以介导其下游效应器线粒体通透性转换孔(mitochondrial permeability transition pore,mPTP)开放,线粒体膜电位去极化,线粒体功能出现障碍,最终导致心肌细胞发生坏死样凋亡。研究证实,通过RIPK3-CaMKII-mPTP通路可以靶向保护心脏免受缺血和氧化应激诱导的坏死样凋亡和心肌重构的影响[16]。抑制CaMKII活性,可阻止RIPK3介导的mPTP开放,说明CaMKII在RIPK3介导的坏死样凋亡中发挥调节信号作用[16]。
3 RIPK3介导的坏死样凋亡在心肌缺血/再灌注损伤中扮演重要角色
坏死样凋亡和心肌梗死之间的联系最早由Luedde等[17]提出,他们在心肌缺血模型中发现,RIPK3表达上调后,依赖RIPK3的坏死样凋亡会导致小鼠心肌缺血后心脏的不良重塑。研究证明,坏死样凋亡是MIRI中坏死性损伤的重要因素之一。阻断坏死样凋亡可以减少约50%的心肌细胞死亡,而泛caspase抑制剂只能减少30%的细胞死亡,这说明坏死样凋亡可能是复氧后心肌细胞死亡的主要方式[18]。
先前的研究将坏死定义为一种不受调控的细胞死亡模式,但目前研究表明,坏死样凋亡是可控的,这为我们提供了通过相应的靶点信号对坏死样凋亡进行调控的新思路。且RIPK3调控的坏死样凋亡在心肌缺血/再灌注损伤下发生的线粒体功能障碍、内质网应激、钙超载、微血管功能障碍、炎症等方面都发挥着一定作用。
3.1 RIPK3介导的坏死样凋亡在心肌线粒体功能障碍中的作用RIPK3介导的坏死样凋亡除了通过破坏质膜发挥作用,RIPK3还会被转移到线粒体膜上,通过激活动力蛋白相关蛋白1(dynamin-related protein 1,Drp1)导致线粒体裂变[19]。Drp1是一种线粒体裂变调节剂,由于缺氧/再灌注(hypoxia/reoxygenation,H/R)损伤,心肌细胞中线粒体分裂调节因子Drp1在第616位丝氨酸上发生磷酸化,并在第637位丝氨酸上发生去磷酸化,从而导致线粒体碎裂且功能紊乱。磷酸甘油酸突变酶5(phosphoglycerate mutase 5,PGAM5)是一种线粒体膜蛋白,在TNF-α诱导的坏死样凋亡模型中负责招募Drp1和Drp1的去磷酸化。在H/R之后,PGAM5表达显著上调,Drp1去磷酸化水平增高,同时线粒体膜电位下降,ROS增加。这说明可以通过抑制PGAM5,进而抑制线粒体分裂蛋白Drp1来减轻H/R诱导的心肌细胞发生坏死样凋亡[19]。
线粒体通透性转换孔(mPTP)已被确定为减少心肌梗死面积,缓解MIRI的重要治疗靶点,它在线粒体形态和功能调控中起着至关重要的作用。mPTP的长时间开放会引发一系列损伤如线粒体功能障碍,最终导致细胞死亡,这是包括心肌损伤在内多种疾病的发病机理[20]。此外,也有研究表明它的开放是细胞发生坏死样凋亡的上游触发因素[21]。
亲环素D(cyclophin-D,Cyp-D)蛋白是位于线粒体中的一种肽基-脯氨酰异构酶,也是mPTP复合体的关键组成部分,它在mPTP的开放调节过程中至关重要。激活的RIPK3上调PGAM5的表达,PGAM5增强Cyp-D的磷酸化作用,Cyp-D继而增加mPTP的开放并介导线粒体断裂增加,从而发生坏死样凋亡[18]。将Cyp-D基因敲除后,小鼠对细胞坏死产生抵抗力,但对细胞凋亡并无抵抗力[20]。综上,RIPK3可以通过不同的通路介导坏死样凋亡,并在线粒体功能障碍导致的心肌损伤中发挥重要作用。
3.2 RIPK3介导的坏死样凋亡参与MIRI中的内质网应激和钙超载研究证明,在心肌梗死后,RIPK3介导的Ca2+/CaMKⅡ激活,进而导致ER应激。同样RIPK3被激活后易位至ER,导致ER应激。ER应激标志物,如78 ku葡萄糖调节蛋白(glucose-regulated protein,GRP78),C / EBP同源蛋白(C/EBP-homologous protein,CHOP)和蛋白激酶R样ER激酶(protein kinase R-like ER kinase,PERK)等表达增加可以表征该过程的发生[21]。
RIPK3还可以促进黄嘌呤氧化酶(XO)和肌醇三磷酸受体(inositol trisphosphate receptor,IP3R)的表达,IP3R是一种位于内质网上的钙通道,负责将钙离子从内质网释放到细胞质,IP3R表达上调后细胞内Ca2+浓度也随之增高[21]。位于内质网表面上的XO是一种负责通过ATP/ADP代谢进行电子传递的氧化酶,在Ca2+调控下它的活性增加,进而导致细胞内的氧化应激和mPTP的开放。所以,在MIRI下,RIPK3能通过内质网应激来上调IP3R表达,提高XO的表达,导致钙超载,并通过ER- Ca2+-XO途径调控ROS的过量产生,进而激活mPTP开放,从而介导MIRI中心肌细胞发生坏死样凋亡[21]。内质网应激不仅参与调控细胞凋亡,也是坏死样凋亡的关键调节因子。
也有研究表明,内质网相关热休克蛋白90(HSP90)是RIPK3的上游激活剂,HSP90从分子伴侣CDC37上解离后可抑制RIPK3介导的坏死样凋亡,这表明HSP90在RIPK3介导的坏死样凋亡中发挥重要作用[22]。
3.3 RIPK3介导的坏死样凋亡与MIRI的心肌微血管功能障碍微血管的通畅性主要受内皮依赖性舒张功能的调节,而内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)的表达在小鼠心脏发生MIRI后显著降低,但敲除RIPK3后,eNOS可恢复到正常水平,且微血管的通畅性也有所改善[18]。此外,经过MIRI后的小鼠心脏,在大多数微血管中会出现线形和不规则的充盈缺损,红细胞易在微血管中积聚,同样,在敲除RIPK3之后,红细胞沉积的概率、微血栓的形成和微血管阻塞的风险均有所降低[18]。
血管内皮钙黏素(vascular endothelial cadherin,VE-cadherin)已被证明是维持血管完整性的关键成分,其表达水平降低会破坏内皮屏障,导致血管通透性增加,炎性细胞浸润。有研究表明MIRI诱发的坏死样凋亡与VE-cadherin表达下降密切相关[23]。此外,RIPK3介导的PGAM5激活和mPTP开放会导致血管舒张剂(eNOS)和血管收缩剂内皮素-1(endothelin-1,ET-1)的失衡,引发微循环障碍,使得“无复流”现象进一步加重。但从敲除RIPK3小鼠的心脏中提取心脏微血管内皮细胞(cardiac microvascular endothelial cells,CMECs),检测RIPK3、p-MLKL和PGAM5的表达,发现均降低到正常水平[18]。
3.4 RIPK3诱导的坏死样凋亡与MIRI中的炎症RIPK3介导MLKL在质膜上执行坏死样凋亡,质膜破裂后,损伤相关分子模式(damage-associated molecular pattern,DAMP)的释放会引发广泛的炎症。而DAMPs一旦释放,便会激活炎症细胞,从而加重炎症和组织损伤。与凋亡相比,坏死样凋亡会破坏质膜的完整性,可能引发更多的炎症反应[14]。此外,许多临床前实验都证明了RIPK3诱导的坏死样凋亡在加重各种器官,包括心脏、肠和皮肤的炎症反应中发挥着不可忽视的作用,且炎症反应反过来又能引起包括坏死样凋亡在内的细胞坏死[11]。
高迁移率族蛋白B1(high mobility group protein B1,HMGB1)是一种DNA结合蛋白,在被分泌到细胞外后,作为一种DAMPs,与晚期糖基化终产物受体(receptor for advanced glycation end products,RAGE)和TLR4结合而发挥促炎活性。HMGB1与TNF-α、IL-1β和IL-6等炎性细胞因子的产生和坏死标志物表达的上调有关。RAGE又会被MIRI上调,故而HMGB1促进了MIRI引起的炎症反应,而炎症反过来又会加剧MIRI。下调HMGB1的表达可以抑制炎症介导的坏死样凋亡,从而保护心肌细胞免受H/R损伤[24]。
羧甲基赖氨酸(Nε-carboxymethyl lysine,CML)是一种晚期糖基化终末物(advanced glycation end product,AGE),由氧化和糖基化反应中的赖氨酸残基进行非酶修饰而产生[25]。AGEs在动脉粥样硬化斑块的形成和糖尿病血管病变中发挥着不可忽视的作用。急性心肌梗塞的患者,特别是在患者发生缺血/再灌注后,CML水平显著升高,可通过增加RIPK3及其下游蛋白的磷酸化来促进RIPK3介导的坏死样凋亡。而RAGE缺乏可以阻断CML对RIPK3介导的心肌坏死样凋亡的作用,缓解CML诱导的MIRI,这表明CML能促进RIPK3介导的心肌坏死样凋亡,并通过RAGE加重MIRI[25]。
4 问题与展望
目前研究已经较为清晰地揭示了坏死样凋亡的分子机制,但许多重要的问题仍未可知。例如,在不同的病理生理条件下,哪些细胞因子、损伤相关分子模式能够刺激坏死样凋亡,以及这些信号如何协调坏死样凋亡;在TNF-α刺激的细胞中,是什么触发了坏死样凋亡激活的关键步骤,即从复合体Ⅰ到复合体Ⅱa/b的转变;RIPK3是如何参与并介导坏死样凋亡、抑制细胞凋亡;MLKL执行坏死样凋亡的分子机制是怎样的?
根据现有的研究,在动物、细胞模型中,RIPK3调控的坏死样凋亡在MIRI的几个方面均起着关键作用,包括心脏炎症、心肌梗死范围扩大、心功能障碍等。但其具体在人体上的效果依然未知。因此,在未来的工作中,仍需要有更多的研究来证明在临床研究中使用RIPK3药理抑制作用的合理性。
猜你喜欢
解放军医学杂志(2021年12期)2022-01-18 03:53:24
现代临床医学(2021年1期)2021-01-26 00:55:52
天津医科大学学报(2019年6期)2019-08-13 07:04:42
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
安徽医科大学学报(2016年12期)2017-01-15 14:21:55
天津科技大学学报(2016年1期)2016-02-28 16:59:45
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
中国当代医药(2015年33期)2015-03-01 02:09:08
现代检验医学杂志(2015年2期)2015-02-06 02:01:01
