
基于CRISPR/Cas9技术构建Plin1基因敲除小鼠模型及表型分析
2022-05-10 12:21燕炯冯晨毅高学坤许祥杨佳敏陈朝阳
生物技术通报 2022年3期
燕炯 冯晨毅 高学坤 许祥 杨佳敏 陈朝阳
(1. 山西医科大学公共卫生学院营养与食品卫生学教研室,太原 030001;2. 山西医科大学实验动物中心,太原 030001)
脂滴(lipid droplet,LD)是大多数真核细胞中储存胞质中性脂质的细胞器。长期以来,LD仅被视为是惰性的脂肪储存库,而如今普遍认为LD是细胞代谢最主要的调节器之一[1]。研究发现,LD表面有上百种蛋白质,这些蛋白质调节着LD的生物发生,并在细胞间通讯、信号传导以及免疫反应等多种生理过程中具有重要作用[2]。
Perilipins是LD表面蛋白家族之一,在哺乳动物中,该家族包含5个成员,其基因序列相似并均具有锚定于LD表面的氨基酸末端序列。围脂滴蛋白(perilipin1,PLIN1)是该家族中最主要的成员,其作为脂肪细胞分化成熟的标志在白色脂肪组织中高度表达,也存在于肝脏,肌肉等组织中[3-5]。近年来的研究显示,PLIN1调节着脂肪细胞的脂质代谢和炎症反应,并与肥胖,糖尿病,高血压等多种疾病的发生发展密切相关[6-10]。然而,目前关于PLIN1在体内的生物学功能尚未完全阐明,仍需要在整体和活体水平进一步研究。本研究通过CRISPR/Cas9基因编辑技术构建Plin1基因敲除小鼠,并对其表型进行初步分析,为后续在体内研究PLIN1作用及机制提供良好的途径与基础。
1 材料与方法
1.1 材料
主要试剂:重组Cas9蛋白购自北京英茂盛业生物科技有限公司;M2培养基、透明质酸酶购自美国Sigma公司;pX330质粒购自美国Addgene公司;限制性核酸内切酶、T4 DNA连接酶购自美国NEB公司;大肠杆菌DH5 α感受态购自北京天根生化科技有限公司;sgRNA体外转录试剂盒购自北京英茂盛业生物科技有限公司;DNA和RNA提取、回收试剂盒及PCR实验相关试剂购自北京天根生化科技有限公司;Perilipin1(ab192716)抗体购自英国Abcam公司,GAPDH(#5174)、β-actin(#3700)抗体购自美国CST公司。
主要仪器:Stepone plus荧光定量PCR仪(美国Applied Biosystems),ON3-99D显微操作系统(日本Olympus公司),MF-900拉针仪(日本Narishige公司)。
实验动物:SPF级别4周龄、15-30 g、100只C57BL/6j小鼠购自赛业(苏州)生物科技有限公司(SCXK(粤)2018-0032)。饲养于山西医科大学动物实验中心(SYXK(晋)2019-0007)。饲养期间各组小鼠自由饮水,D1245高脂饲料和D12450B小鼠生长维持饲料购自北京斯贝福生物技术有限公司。饲养环境:昼夜各半循环照明,湿度恒定,温度控制在24-28℃。所有操作均符合实验伦理学要求(审批号 :ACU20-I001)。
1.2 方法
1.2.1 sgRNA的设计与筛选 在NCBI中查询小鼠Plin1基因基本信息,利用在线sgRNA设计工具(http://crispr.mit.edu/),针对Plin1基因2号外显子前后序列设计sgRNA,敲除策略如图1所示。脱靶位点、基因数和发生错配的可能性大小进行评估,筛选出2条评分较高sgRNA,sgRNA及其PAM序列见表1。以设计好的sgRNA为模板互补配对相应的反义链,并在5′端增加黏性末端后交北京微旋基因技术有限公司合成。

表1 sgRNA序列Table 1 sgRNA sequence
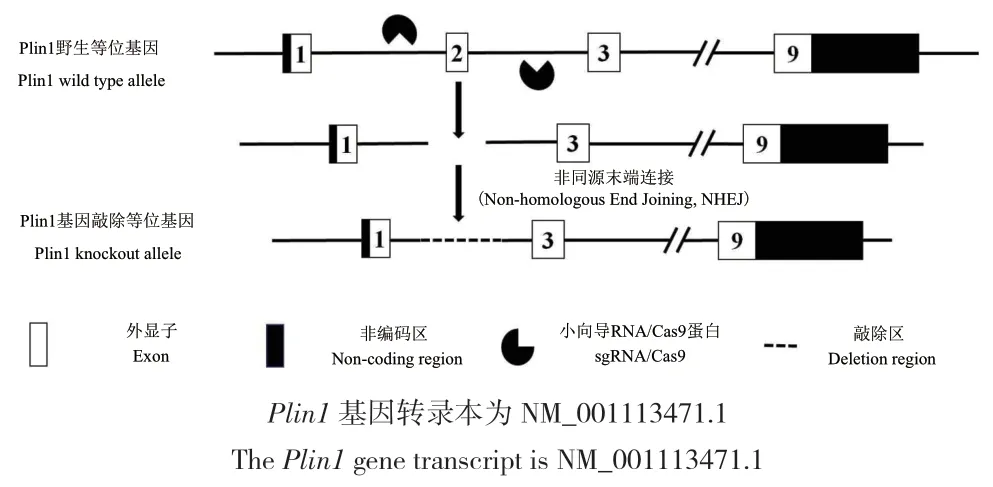
图1 sgRNA敲除Plin1基因2号外显子的示意图Fig.1 Schematic diagram of sgRNA knocking out exon 2 of Plin1 gene
1.2.2 sgRNA表达载体的构建与体外转录 将sgRNA双链退火磷酸化后稀释至1 μmol/L,同时利用限制性内切酶Bbs I切割pX330骨架质粒载体使其线性化,DNA回收试剂盒回收线性化质粒载体后分别与2条sgRNA双链在含有T4 DNA连接酶的体系中进行连接,16℃过夜。构建好的质粒载体转化至DH5α菌群中,常规培养2 d后提取质粒DNA进行ApaL I+Nco I双酶切法鉴定并进行测序检测。载体构建成功按照试剂盒说明书设计引物进行PCR获得sgRNA体外转录模板,引物序列为:SG-F1:TTAATACGACTCACTATAGGGTTGGAGGGCGAATGTTACATGTTTTAGAGCTAGAAATAG,SG-F2:TTAATACGACTCACTATAGGGTTGGAGGGCGAATGTTACATGTTTTAGAGCTAGAAATAG,SG-R:AAAAAAAGCACCGACTCGGTGCCACTTTTTC,利用sgRNA转录模板体外转录后检测转录产物的大小及完整性,并使用RNA纯化试剂盒进行纯化,通过微孔板分光光度计测定转录产物的的浓度和纯度(A260/A280值)。
1.2.3 受精卵的获得与显微注射 5-10 IU的孕马血清和人绒毛膜促性腺激素促使雌性小鼠超数排卵后与雄性小鼠合笼,次日挑取见栓雌鼠剪取双侧输卵管,置于含有3%透明质酸酶液滴的培养皿中,用精细镊子撕破壶腹部使受精卵流出,短暂消化至受精卵散开后用M2培养基清洗3次,移入一个含有M2培养基液滴的新培养皿中并加入矿物油,置于显微注射操作系统中。将sgRNA与Cas9蛋白混合后调整至适宜浓度,通过注射针向受精卵原核中注射1-2 pL,注射完毕后,挑取存活的受精卵移入M16培养基中,5% CO2、37℃条件下培养2 h。
1.2.4 Plin1基因小鼠的获得及基因型鉴定 将经显微注射后培养存活的受精卵通过输卵管伞端移植入假孕雌鼠体内,19-21 d后小鼠出生剪尾提取DNA,PCR及测序进行基因型鉴定筛选,获得F0代小鼠;F0代小鼠与异性野生型小鼠交配,后代基因型鉴定获得F1代杂合型基因敲除小鼠;F1代杂合小鼠雌雄近交,后代基因型鉴定获得F2代纯合型基因敲除小鼠。针对敲除区域设计了F、R1和R2三条引物鉴定基因敲除及野生型小鼠,鉴定策略如图2所示,引物序列见表2所示。

图2 Plin1基因小鼠敲除基因型鉴定的策略Fig.2 Strategies for genotype identification of Plin1 knockout mice

表2 基因型鉴定的引物序列Table 2 Primer sequence for genotyping
1.2.5 小鼠一般体格指标测定 常规喂养4周后,以同窝别杂合及野生型小鼠作为对照,测量小鼠空腹体重、体长并记录每周摄食量。
1.2.6 qRT-PCR检测小鼠PLIN1 mRNA水平 提取小鼠肝脏、骨骼肌及脂肪组织中总RNA,去除gDNA后加入反转录体系,95℃加热3 min获得cDNA。按照2× M5 HiPer SYBR Premix EsTaq(with Tli RNaseH)使用说明书以及Light Cycler 96 System操作说明配置反应体系,PCR反应条件为95℃ 预变 性 30 s后,95℃ 5 s,60℃ 20 s,65℃ 15 s,40个循环条件PCR。引物序列F:5′-CAACCCTGCTGGATGGAGACCT-3′,R :5′-GGTGGGCTTCTTTGGTGCTGTT-3′,以小鼠gapdh作为内参基因,引物序列F :5′-AGGTCGGTGTGAACGGATTTG-3′,R :5′-TGTAGACCATGTAGTTGAGGTCA-3′。
1.2.7 Western-blot检测小鼠PLIN1蛋白表达水平 提取小鼠肝脏、骨骼肌及脂肪组织中总蛋白并定量蛋白浓度,100℃金属浴变性后制备SDS-PAGE凝胶。80 V,15 min;100 V,60 min电泳后;120 min,220 mA恒流转膜。5%的脱脂奶粉封闭后一抗(稀释比例为1∶1 000)孵育,4℃过夜,室温二抗孵育120 min。NC膜漂洗数次后滴加ECL plus,于成像系统下曝光。Image J软件检测各蛋白条带灰度值,以GAPDH或β-actin条带进行校正。
1.2.8 数据分析 本次研究所有数据均采用SPSS 22.0软件进行分析,各组数据采用描述,组间比较采用one-way ANOVA,P < 0.05具有统计学意义。
2 结果
2.1 sgRNA表达载体鉴定及体外转录
如 图 3-A所 示,ApaL I( 第 6 376、6 837和8 119位置)+Nco I(第654和1 181位置)酶切sgRNA表达载体后,获得了5个预期的大小的片段(497、527、959、1 246和5 195 bp);如图3-B所示,经基因测序后,sgRNA-Plin1-1、2插入在pX330 gRNA scafflod位置,表明sgRNA表达载体Plin1-1、2构建成功;sgRNA表达载体完全体外转录后,获得sgRNA体外转录片段完整,如图3-C所示,大小在100-200 bp之间,与预期结果一致。对转录产物进行纯化回收,浓度均大于5 000 ng/L,A260/A280比值为2.03±0.06,表明体外转录sgRNA成功。
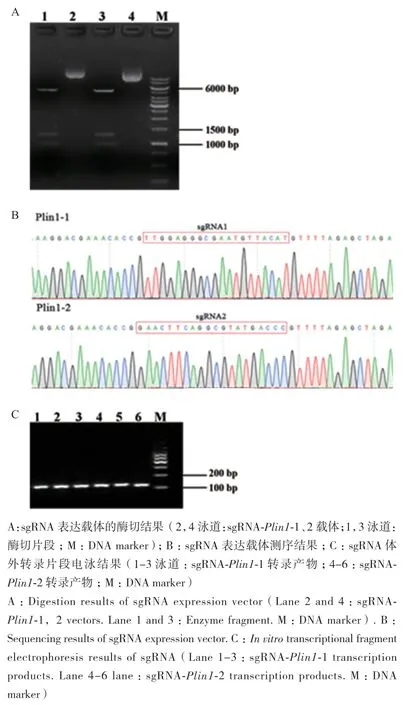
图3 sgRNA表达载体构建与鉴定Fig.3 Construction and identification of sgRNA expression vector
2.2 纯合型Plin1基因敲除小鼠的获得
本研究通过显微注射受精卵数共213枚,可移植卵数为118枚,分别移植入4只假孕雌鼠体内;出生小鼠24只,出生率为20.3%;其中F0阳性鼠12只(5♂7♀),阳性率为50.0%,F0代阳性小鼠Plin1基因缺失740 bp左右,PCR电泳结果如图4-A所示;选择8号F0雄性小鼠测序后与8周龄野生型雌鼠交配,获得F1代小鼠13只(4♂9♀);F1代杂合型小鼠雌雄近交获得F2代纯合型小鼠2只(1♂1♀)、杂合型小鼠13只(6♂7♀)及野生型小鼠2只(2♀);F1代小鼠PCR电泳结果如图4-B-C所示;F0代8号小鼠和部分F1代小鼠基因测序结果如图4-D所示;F2代小鼠基因型的鉴定结果如图5所示。
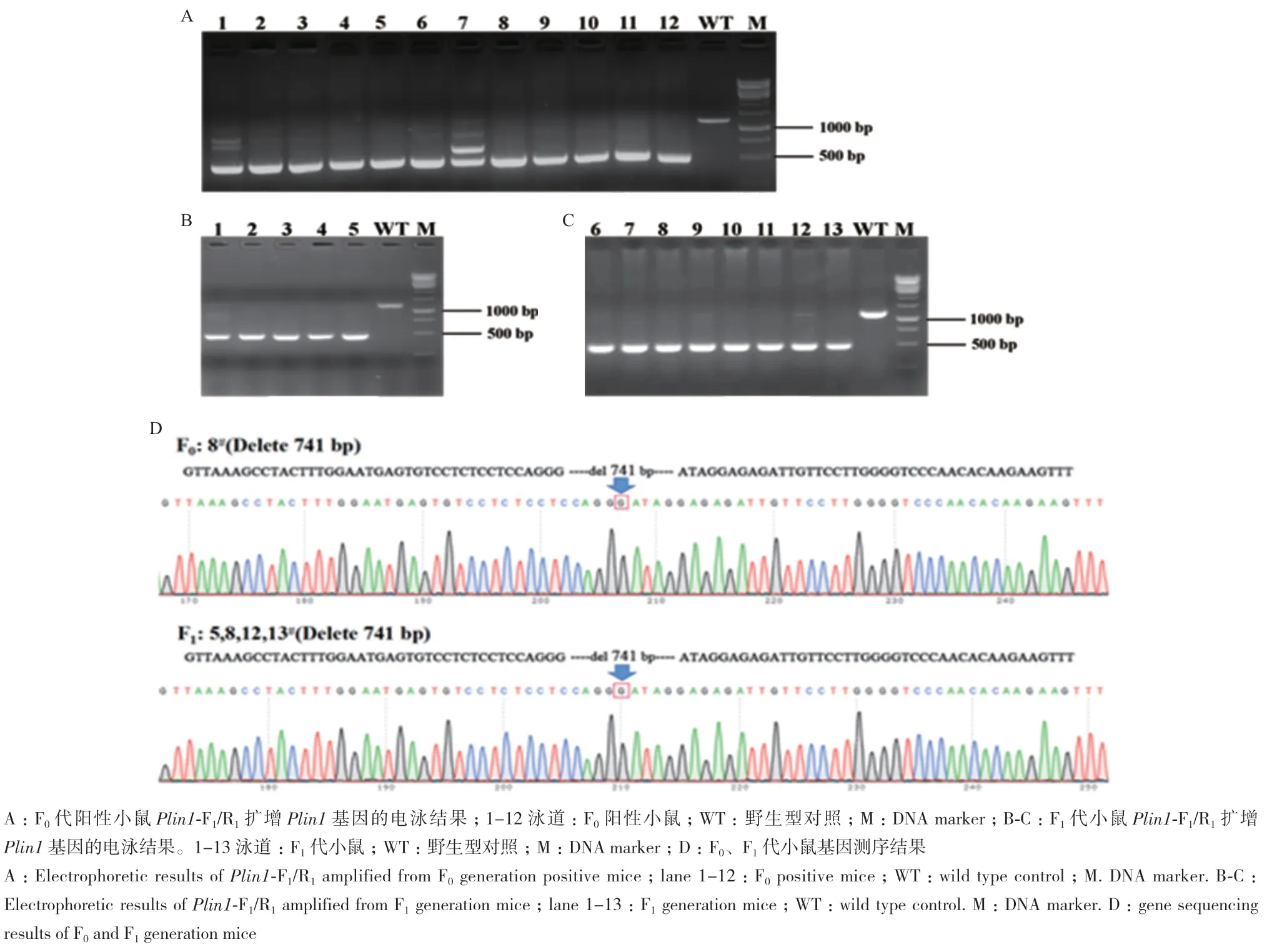
图4 F0和F1代小鼠基因型鉴定Fig.4 Genotype identification of F0 and F1 generation mice

图5 F2代小鼠基因型鉴定Fig.5 Genotype identification of F2 generation mice
2.3 纯合Plin1基因敲除小鼠一般指标
常规饲料喂养4周后,与同周龄野生型小鼠相比,F2代杂合型Plin1基因敲除小鼠体重、体长及每周摄食量并无明显变化,而F2代12,13号小鼠近交获得的F3代纯合型Plin1基因敲除小鼠体重显著降低,差异有显著性(P < 0.05);体长与每周摄食量差异无显著性表达(表3)。

表3 各组小鼠体格参数Table 3 Physical parameters of mice in each group
2.4 纯合型Plin1基因敲除小鼠PLIN1 mRNA水平
取F3代纯合型、F2代杂合型Plin1基因敲除小鼠及同窝别的野生型小鼠各2只,Q-RT-PCR检测小鼠肝脏、骨骼肌、白色脂肪组织中PLIN1 mRNA水平,如图6所示,相比于野生型小鼠Plin1基因敲除小鼠在各组织中PLIN1 mRNA水平显著降低(P < 0.05),杂合型Plin1基因敲除小鼠PLIN1 mRNA水平降低约49.3%(a:肝脏48.0%、b:骨骼肌51.1%、c:白色脂48.7%),纯合型Plin1基因敲除小鼠基本无PLIN1 mRNA表达。

图6 小鼠各组织中PLIN1 mRNA的相对表达量Fig.6 Relative expressions of PLIN1 mRNA in the various tissues of mice
2.5 纯合型Plin1基因敲除小鼠PLIN1蛋白表达水平
Western-blot检测小鼠各组织中PLIN1的蛋白表水平,如图7所示,相比于野生型小鼠,Plin1基因敲除小鼠在各组织中PLIN1蛋白的表达水平显著降低(P < 0.05),杂合型Plin1基因敲除小鼠PLIN1蛋白水平降低了46.5%(A:肝脏41.1%、B:骨骼肌60.9%、C:白色脂肪37.5%),纯合型Plin1基因敲除小鼠基本无PLIN1蛋白表达。
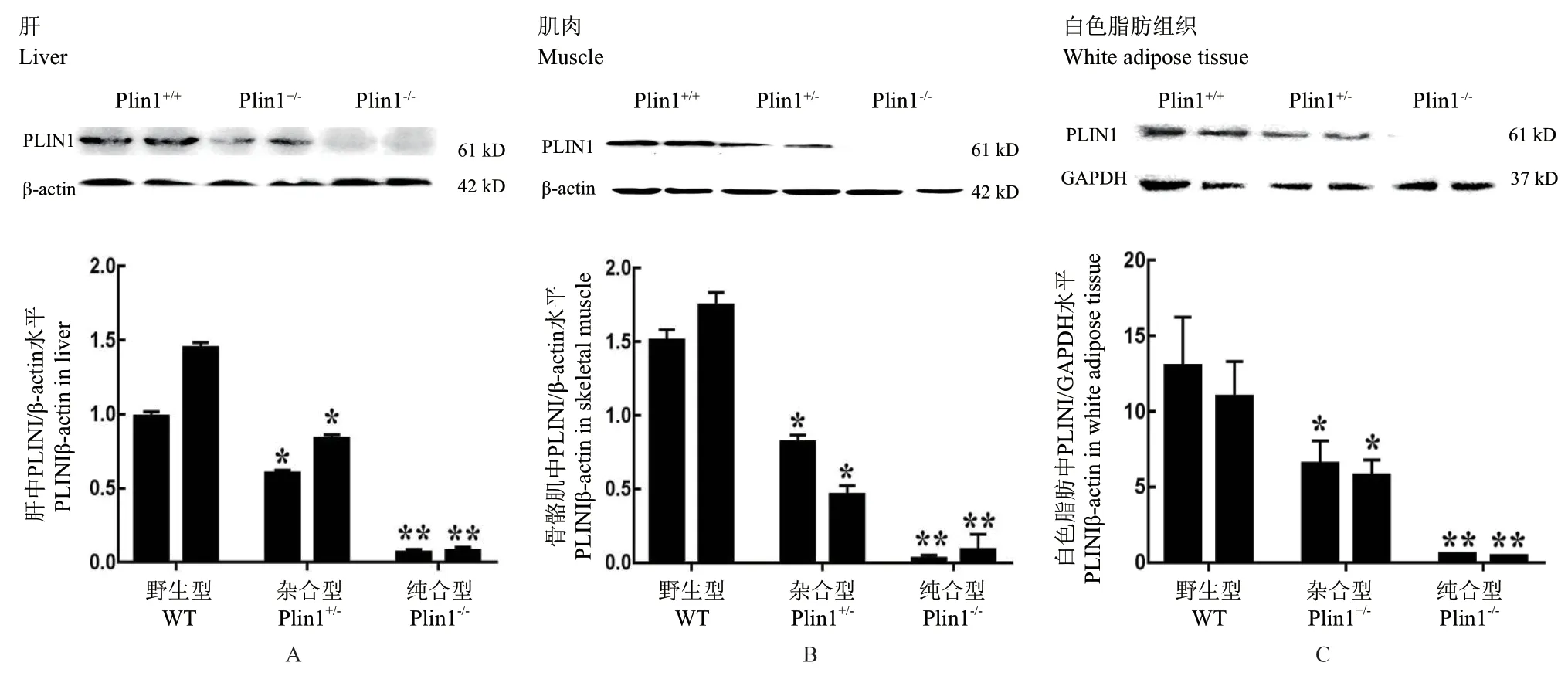
图7 小鼠各组织中PLIN1的蛋白表达水平Fig.7 Protein expressions of PLIN1 in the various tissues of mice
3 讨论
人类Plin1基因单拷贝编码PLIN1,位于15号染色体(15q26.1)的长臂上,基因编号5346,其DNA长度大于15 kb,包含9个外显子和8个内含子(https://www.ncbi.nlm.nih.gov/gene/5346)[10]。 研究发现,Plin1基因与人群肥胖的发生密切相关,在2005年已被列为肥胖相关基因之一[11]。然而,关于Plin1基因与肥胖发生关系的研究大多集中在细胞及组织水平,缺乏从整体动物水平着手的相关研究。鉴于目前通过基因敲除建立稳定的实验动物模型已成为研究基因功能的金标准,因此我们构建了Plin1基因敲除小鼠模型,旨在进一步在动物体内研究该基因的功能。
CRISPR/Cas9作为第三代基因编辑技术,得益于其操作简单、高效率和低成本的优势在基因编辑领域被广泛应用[12-13]。CRISPR/Cas9系统组分包括Cas9蛋白和sgRNA,通过sgRNA识别目的基因并引导Cas9蛋白切割,在靶点位置造成双链断裂,之后再在DNA损伤修复机制的作用下实现移码突变以达到基因敲除的目的[14]。利用显微注射等技术将靶向目的基因的CRISPR/Cas9系统组分递送至受精卵中后再对其进行胚胎移植是目前获得基因敲除小鼠最便捷的方法[15]。在前期研究中,我们已经成功构建了Plin1基因敲除载体并在细胞内外验证了其切割活性[16]。而在本次研究中,我们基于CRISPR/Cas9技术在Plin1基因2号外显子前后序列设计一对sgRNA,通过构建sgRNA表达载体和体外转录,获得了预期的sgRNA片段,之后通过与Cas9蛋白混合并利用显微注射的方式递送至小鼠受精卵细胞中,PCR和基因测序结果表明成功敲除了包含Plin1基因2号外显子序列在内741 bp片段。之后通过F0代小鼠和野生型小鼠的杂交及子代小鼠的不断近交,在敲除区域中设计引物,通过PCR鉴定筛选得到了纯合型Plin1基因敲除小鼠。
PLIN1是一种相对分子质量为56-62 kD的LD表面蛋白,由522个氨基酸组成,高表达于白色脂肪组织中,也存在于在肝脏、肌肉等组织中[17]。研究发现,PLIN1在LD表面充当蛋白屏障并与比较基因 鉴 定 58(comparative gene identification-58,CGI-58)结合,在基础状态下避免LD中的中性脂质被无效降解,在激素刺激作用介导激素敏感性脂肪酶(hormone-sensitive lipase,HSL)至脂滴表面的易位,同时释放CGI-58活化脂肪甘油三酯脂肪酶(adipose triglyceride lipase,ATGL)促进脂解[18-19]。课题组前期利用RNAi技术已证实沉默Plin1基因下调了3T3-L1脂肪细胞中PLIN1的表达后,促进了脂肪细胞脂解,降低了胞内TG的水平[20-21]。有研究也发现,Plin1基因敲除和过表达小鼠对高脂饮食诱导的肥胖均具有抵抗作用[22-23]。本次研究结果显示,敲除Plin1基因2号外显子在内741 bp后实现了移码突变,杂合及纯合型Plin1基因敲除小鼠各组织中PLIN1 mRNA和蛋白表达显著下调,纯合型Plin1基因敲除小鼠基本无PLIN1 mRNA和蛋白表达。通过常规饲料喂养4周后初步分析其表型发现,相比于野生及杂合型小鼠,纯合型Plin1基因敲除小鼠体重明显降低,这可能是由于PLIN1失活后,LD表面失去了蛋白屏障,脂解酶易于水解其内的脂质,LD的体积缩小,从而导致脂肪组织萎缩所引起的,但具体作用及机制尚待进一步探究。
综上所述,本研究基于CRISPR/Cas9技术成功构建了纯合型Plin1基因敲除小鼠,初步表型分析发现纯合型Plin1基因敲除小鼠体重相比于野生型小鼠显著降低,较为瘦小。这些结果为在体内进一步研究PLIN1的功能及其与肥胖发生的关系奠定了基础。
4 结论
本研究基于CRISPR/Cas9技术成功构建了Plin1基因敲除小鼠模型,并且发现Plin1基因敲除小鼠体型较为瘦小。这为后续探究PLIN1的功能及其与肥胖的关系提供了技术支持和参考依据。
猜你喜欢
华人时刊(2022年9期)2022-09-06
种子(2021年3期)2021-04-12
华人时刊(2020年15期)2020-12-14
中国生育健康杂志(2018年6期)2018-11-13
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
作物研究(2014年6期)2014-03-01
中国火炬(2013年11期)2013-07-25
中国火炬(2013年10期)2013-07-24
中国糖料(2013年1期)2013-01-22
