磷酸二酯酶9A在心肌细胞缺血缺氧再灌注损伤的表达变化及意义
2022-04-26 10:34李梦豪李幼奇邓焕堂罗艳芳农盛雄谢雄伟刘锦光
医学研究生学报 2022年3期
申 健,李梦豪,李幼奇,邓焕堂,张 宇,罗艳芳,农盛雄,谢雄伟,刘锦光
0 引 言
急性心肌梗死及心力衰竭已成为世界性的公共卫生难题。及时再灌注治疗能显著改善心肌梗死患者的预后[1],但仍有相当部分患者无法及时得到有效治疗,且部分心肌梗死患者即使得到及时再灌注治疗,仍然难以避免并发心力衰竭,后者被认为与缺血再灌注损伤、细胞凋亡等机制有关[2-3]。最近研究表明,新发现的磷酸二酯酶9A(phosphodiesterase 9A, PDE9A)在心力衰竭或压力负荷刺激下上调并导致心肌重构,且其导致心肌重构的细胞信号转导途径不依赖一氧化氮信号通路,而是由脑利钠肽偶联的环磷酸鸟苷(cyclic guanosine monophosphate, cGMP)所介导[4-5]。最近的研究主要集中在PDE9A与阿尔茨海默病、心肌肥厚、动脉粥样硬化相关机制方面[6-8],但对于其与急性心肌梗死、缺血再灌注损伤及随后的心肌重构及心力衰竭的关系方面未作探讨。本研究拟探讨PDE9A在心肌细胞缺血缺氧再灌注损伤的表达变化,以及干预其表达对下游信号和细胞凋亡的影响,探讨心肌梗死后心肌重构和心力衰竭的新机制,以期为相关药物的研发开辟一条新途径。
1 材料与方法
1.1 实验动物与材料新生1~3d的SD大鼠乳鼠,购自南京医科大学,动物许可证号:SCXK(苏)2021-0001。动物实验按照SPF级要求饲养,室内温度20~26 ℃,相对湿度40%~70%,光照12 h明暗交替,自由进食进水。胎牛血清和 DMEM 高糖培养基购自美国Hyclone公司,胶原酶 I、Sil 及 Iso 购自美国Sigma 公司, CCK-8、乳酸脱氢酶(lactate dehydrogenase, LDH)细胞毒性试剂盒、BCA 蛋白质测定试剂盒及辣根过氧化物酶标记山羊抗兔 IgG 均购自碧云天生物有限公司,annexinV-FITC/PI 凋亡检测试剂盒购自美国 BD公司,兔抗葡萄糖调节蛋白78(glucose regulatory protein78, GRP78)、兔抗 CCAAT增强子结合蛋白同源蛋白(CCAATenhancerbindingproteinhomologousprotein, CHOP)及兔抗 β-actin 购自美国 CST 公司,PVDF 膜和 ECL 发光液购自美国Millipore公司,全波长酶标仪购自美国BioTek,流式细胞仪购自美国 BD 公司,电泳仪、垂直电泳槽、转膜槽以及凝胶成像系统购自美国 Bio-Red 公司,BAY 73-6691购自德国拜耳公司。
1.2原代心肌细胞的培养和鉴定取新生1~3 d的的SD大鼠乳鼠用75%的乙醇消毒后,在无菌条件下迅速取出心室肌,用4℃预冷的PBS液清洗3次,去除血污,剪成1 mm3的碎块,加0.25%胰蛋白酶消化液在37℃水浴锅中磁力搅拌并分次消化(5 min/次)。1000r/ min离心10 min,收集心肌细胞。加入适量RPMI 1640培养基悬浮,放入37℃,5%CO2培养箱培养。4h后,轻轻吹打吸出悬浮的细胞悬液(去除非心肌细胞),计数并调整细胞浓度3×105个/mL,置于37℃ 5% CO2培养箱培养。显微镜下观察心肌细胞形态,采用α-SAM免疫荧光鉴定心肌细胞,以台盼蓝染色法检测心肌细胞成活率。
1.3氧糖剥夺/再灌注法(oxygen glucose deprivation/reperfusion, OGD/R)模型构建采用培养3 d的乳鼠心肌细胞,换用高纯氮气饱和的无糖MEM培养基缺血缺氧作用2h(I/R)后,再加入适量RPMI 1640培养基,放入37 ℃,5%CO2培养箱培养12 h复氧,构建心肌OGD/R模型。对照组细胞正常培养。
1.4RT-qPCR检测PDE9的相对表达量及荧光定量PCR。引物由武汉金开瑞公司合成。序列见表1。
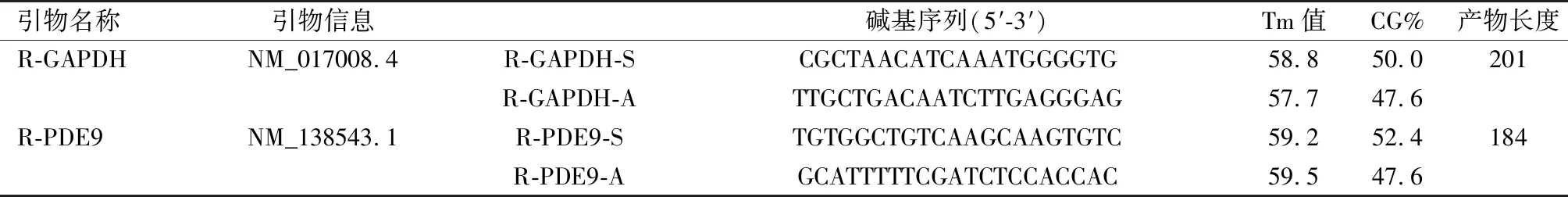
表 1 RT-qPCR检测PDE9的引物序列
1.5Western blot检测PDE9蛋白含量Western blot检测对照组与OGD/R组中PDE9A的表达。
1.6实验分组及处理分为对照组(心肌细胞不干预)、OGD/R组(构建心肌OGD/R模型)、siPDE9A组(构建PDE9A的干扰质粒转染入OGD/R细胞)、sGC组(正常细胞加入sGC抑制剂)、sGC+siPDE9A组(PDE9A的干扰质粒转染入OGD/R细胞,并加sGC抑制剂)、sGC+rGC+siPDE9A组(sGC抑制剂+rGC抑制剂+siPDE9A)。采用Western blot检测PDE9A、cGMP的表达,流式细胞术检测细胞凋亡水平。
1.7应用PDE9抑制剂培养原代乳鼠的心肌细胞鉴定后,采用OGD/R构建心肌细胞缺糖缺氧的模型;向OGD/R的心肌细胞中加入PDE9的抑制剂BAY 73-6691,共孵育一定时间,并按不同时间分为30min组、60min组、120min组,180min组及240min组,采用CCK8技术观察细胞活性,流式细胞检测细胞凋亡情况,ATP检测细胞线粒体活性,以及采用Western blot检测PDE9A 和cGMP及其下游相关蛋白(cGMP、Bax、Caspase-3、CREB、Caspase-9和Bcl-2)的级联变化。

2 结 果
2.1 心肌细胞的鉴定及成活率检测显微镜下显示,心肌细胞呈圆形,单个细胞可出现自发性波动。α-SAM在心肌细胞表达阳性率超过98%,台盼蓝染色显示心肌细胞的成活率超过90%。见图1。
2.2RT-qPCR检测对照组与OGD/R组中PDE9A的表达OGD/R组PDE9A表达量(1.0±0.21)显著高于对照组(2.13±0.26),差异有统计学意义(P<0.01)。
2.3Western blot检测对照组与OGD/R组中PDE9A的表达Western blot显示,OGD/R组中PDE9A蛋白表达量显著高于对照组[(0.84±0.01)vs(0.38±0.03),P<0.001]。见图2。
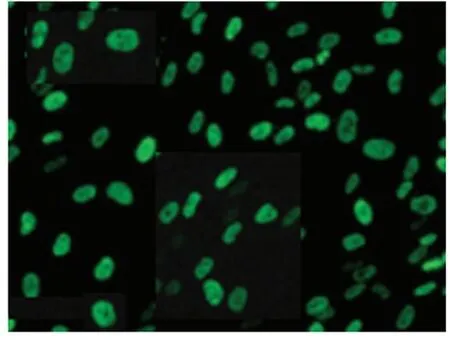
图 1 α-SAM在心肌细胞表达(×100)
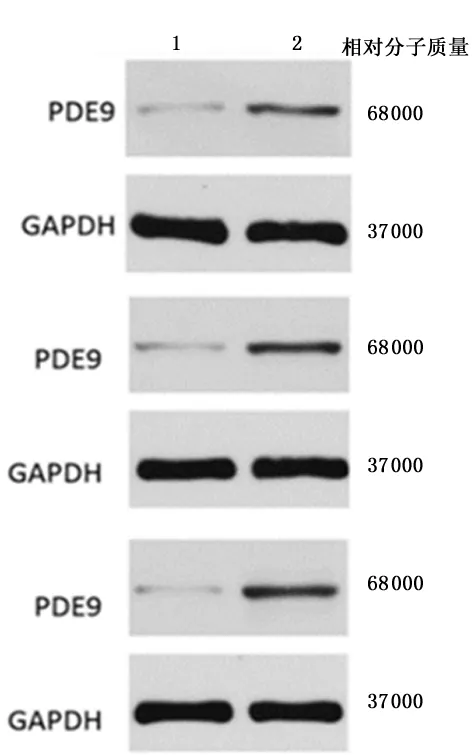
1:对照组; 2:OGD/R组
2.4Western blot检测转染siPDE9A后PDE9A蛋白的表达与对照组(1.09±0.11)比较,转染siPDE9至大鼠心肌细胞时,siPDE9A组的PDE9A蛋白(0.36±0.03)被显著抑制(P<0.001)。见图3。
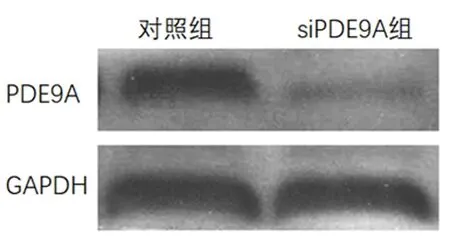
图 3 转染siPDE9A后Western blot检测PDE9A蛋白表达及抑制情况
2.5PDE9A、cGMP蛋白表达情况与对照组比较,OGD/R组cGMP蛋白表达显著抑制,PDE9A蛋白表达显著增加(P<0.001);与OGD/R组比较,sGC组的cGMP蛋白表达显著抑制;siPDE9A组、siPDE9A+sGC组、 siPDE9A+sGC+rGC组的PDE9A蛋白表达显著抑制(P<0.001), cGMP蛋白表达显著增加(P<0.001)。见表2。
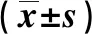
表 2 Western blot检测PDE9A、cGMP蛋白表达情况
2.6流式检测细胞凋亡流式细胞检测结果提示,与OGD/R组比较,对照组、siPDE9A 组、siPDE9A+sGC组的细胞凋亡率显著抑制。见图4。
2.7Western blot检测PDE9A、cGMP、Bax、Caspase-3、CREB、Caspase-9、Bcl-2蛋白表达情况与对照组比较,OGD/R组的PDE9A、Bax、Caspase-3、CREB、Caspase-9蛋白表达显著增加(P<0.01), Bcl-2、cGMP蛋白表达显著降低(P<0.01);与OGD/R组比较, 60 min组、120 min组、180 min组的PDE9A、Bax、Caspase-3、Caspase-9蛋白表达显著降低(P<0.01),Bcl-2蛋白表达显著增加(P<0.01);30 min组、 60 min组、120 min组的CREB、cGMP蛋白表达显著增加(P<0.01)。见图5,表3。
2.8流式检测细胞凋亡流式细胞检测结果表明,与OGD/R组比较,对照组、60min组、120min组、180min组的细胞凋亡率显著抑制(P<0.05)。见图6。
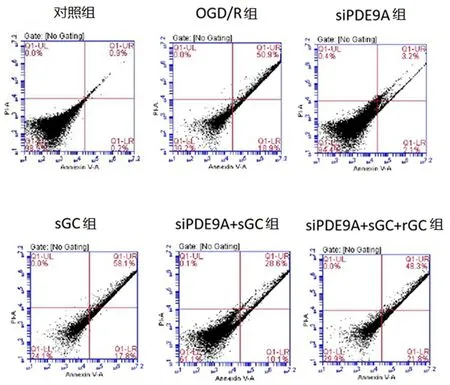
图 4 流式检测各组细胞凋亡情况
1:对照组; 2:OGD/R组; 3:30 min组; 4:60 min组; 5:120 min组; 6:180 min组; 7:240 min组

表 3 PDE9A、cGMP、Bax、Caspase-3、CREB、Caspase-9、Bcl-2蛋白表达情况
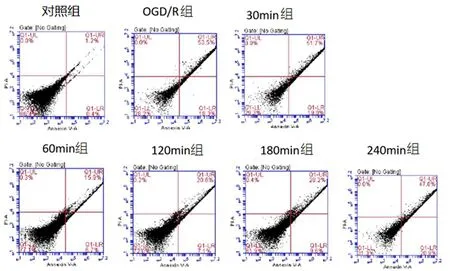
图 6 应用PDE9抑制剂后流式检测细胞凋亡
2.9CCK-8检测细胞增殖CCK-8检测结果表明,与OGD/R组比较,对照组、60 min组、120 min组、180 min组、240 min组的细胞增殖显著上升(P<0.05)。见图7。
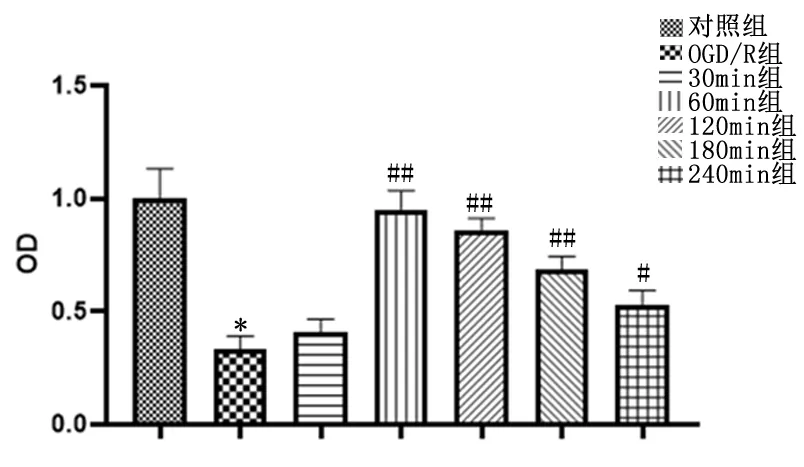
与对照组比较,*P<0.05; 与OGD/R组比较,#P<0.05,##P<0.01
2.10ATP检测线粒体活性ATP检测结果表明,与OGD/R组比较,对照组、60 min组、120 min组、180 min组的ATP含量显著上升(P<0.05)。见图8。
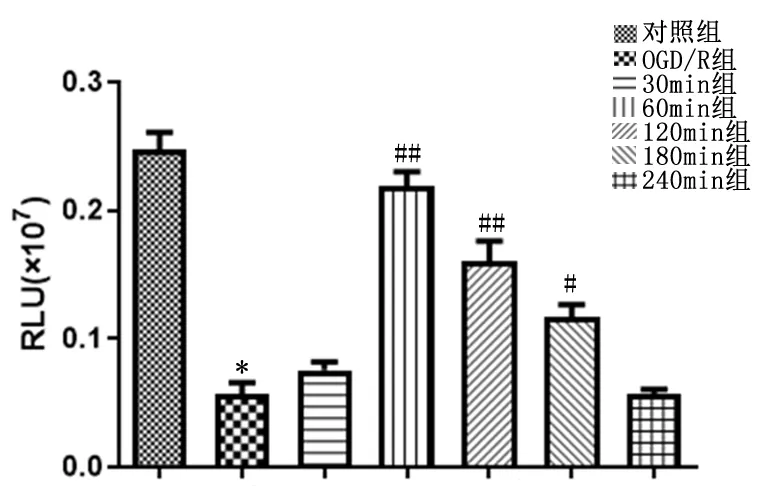
与对照组比较,*P<0.01; 与OGD/R组比较,#P<0.05,##P<0.01
3 讨 论
PDE是一类可水解细胞内第二信使cAMP及cGMP的酶类。PDE通过即时地调整控制细胞内第二信使的含量从而参与调控了多种物质代谢和能量代谢过程。PDE迄今共发现了11种不同的同工酶家族,其中各家族又包含不同的亚型,这些酶特异性分布于不同的组织器官并作用于不同条件下的环磷酸腺苷(cyclicadenosinemonophosphate, cAMP)或cGMP的水解,其表达量、调节方式以及对于抑制剂的敏感程度均不同。这些特点使得PDE成为药物研究的热门靶点,能在有效治疗的基础上最大的程度地减少药物在其他非病灶组织的副作用[9-10]。
既往研究发现,促进cGMP合成或通过阻断PDE5A抑制cGMP的降解,这样能强化心脏细胞内cGMP的作用,对心血管疾病和肾脏疾病有一定治疗作用[11-13]。但是这种治疗本身会诱导细胞PDE表达增加,出现药物耐受。尽管PDE5A能调节NO诱导的cGMP水平,但在心力衰竭患者中,由于NOS可用性受损,故抑制PDE5的疗效受到一定限制[4]。综上所述,抑制PDE5A可能并非心脏疾病理想的治疗靶点[14],临床药物的使用也印证了这一点,如PDE5抑制剂西地那非、他达拉非等药物,主要用于治疗肺动脉高压而非心力衰竭[15]。
最近有研究表明,新发现的PDE9A基因在心力衰竭或压力负荷刺激下上调并导致心肌重构,还在动脉粥样硬化性心脏病中上调表达,且其细胞信号转导途径不依赖一氧化氮信号通路(nitric oxide signaling pathway, NOS)而是由脑利钠肽偶联的cGMP所介导,对NO诱导的cGMP无明显作用[4-5],但其在缺血再灌注损伤及缺血性心力衰竭中的作用尚不明确。
本研究通过构建SD乳鼠心肌细胞的OGD/R模型发现,PDE9A在OGD/R组中的表达量显著上调;转染干扰PDE9A或使用PDE9抑制剂能够降低其细胞凋亡或促进细胞增殖、降低细胞内线粒体损伤。本研究还发现,OGD/R模型cGMP水平较对照组显著降低,在OGD/R基础上再抑制NOS但不抑制PDE9A(sGC组),cGMP水平最低。抑制PDE9A后(siPDE9A组)cGMP水平有所上升,且同时抑制PDE9A与NOS后(siPDE9A+sGC组、siPDE9A+sGC+rGC组),细胞内cGMP水平较单纯抑制NOS升高;而且本研究还提示,单纯抑制NOS后(sGC组),PDE9A蛋白表达并无显著变化,以上说明抑制PDE9A可上调cGMP,且其细胞信号转导通路不完全通过NOS-cGMP途径。本研究通过多方面验证,通过干扰PDE9A表达或直接应用抑制剂抑制PDE9A均能降低因缺血缺氧再灌注损伤所致的细胞凋亡,其中均伴随着cGMP水平的增加,故推测抑制PDE9A的抗凋亡作用机制可能与cGMP水平升高有关。
本研究尚有不足之处,尚未在动物水平上验证PDE9A的变化及功能,针对PDE9A上下游的具体信号通路仍有待进一步分析。
综上,PDE9A可能参与急性心肌梗死及其缺血再灌注损伤过程,类比其在心肌肥厚、慢性心力衰竭进程中的功能,本实验推测PDE9A在缺血性心力衰竭进程中的上调使得心肌细胞内cGMP减少,失去了其对心肌细胞的保护作用,进而参与心肌重构。PDE9A表达不完全通过NOS-cGMP途径,使得针对该靶点的干预可能较抑制其它类型的PDE更有效。该项研究继续深入,如使用全转录组测序、生物信息学分析及靶点验证,分析PDE9A在心肌梗死后心力衰竭进程中的上下游调控机制,可能将进一步阐明心肌梗死后心肌重构和心力衰竭的发生发展机制,特别是PDE9A在其中的重要角色和相关机制,为干预心衰进程的治疗手段和相关药物的研发开辟一条新的途径。
猜你喜欢
现代临床医学(2022年3期)2022-06-06
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
中国典型病例大全(2022年10期)2022-05-10
医学概论(2022年4期)2022-04-24
中西医结合心脑血管病杂志(2022年4期)2022-03-11
中西医结合心脑血管病杂志(2022年2期)2022-02-15
保健与生活(2021年11期)2021-06-10
家庭医药(2019年8期)2019-08-27
体育科学(2018年12期)2019-01-04