
Effect of Different DNA Activator Structures on the trans-Cleavage Activity of Cas12a
2022-04-22 03:37FEIXinruiLIUXiaolingLIUChenghui
分析测试学报 2022年4期
FEI Xin-rui,LIU Xiao-ling,LIU Cheng-hui
(Key Laboratory of Applied Surface and Colloid Chemistry,Ministry of Education;Key Laboratory of Analytical Chemistry for Life Science of Shaanxi Province,School of Chemistry&Chemical Engineering,Shaanxi Normal University,Xi'an 710119,China)
Abstract:The t rans-cleavage activity of CRISPR-Cas12a system could be activated after it recognizes the specific DNA activator sequences,which paves the foundation not only for direct target DNA detection but also for expanding indirect biosensing of various biomolecules.However,both double-strand DNA(dsDNA)and single-strand DNA(ss-DNA)activators with varying structures are employed in the existing literatures,and there still lacks systematic guiding principles for Cas12a activator design.Herein,the impact of DNA activator structures on the t rans-cleavage activity of LbaCas12a is systematically studied by monitoring the fluorescence signal generated under the guidance of the same crRNA.According to a series of comparison,the following conclusions have been drawn for the sequence design of DNA activators.(1)protospacer-adjacent motif(PAM)site helps LbaCas12a to target the dsDNA activators and the ssDNA activators with higher efficiency.(2)lacking the sequence fragment in the proximal region of PAM will reduce the efficiency of Cas12a-crRNA in positioning the activator.(3)pre-deletion of the fragment adjacent to the crRNA-pairing sequence in the PAM-distal end is conducive to the LbaCas12a trans-cleavage activity.(4)ssDNA activators generally have better performance in activating the trans-cleavage activity of LbaCas12a than dsDNA activators due to the omission of dsDNA unzipping.According to these findings,an efficient LbaCas12a-preferred activator structure is recommended,which could yield 3.7 times higher fluorescence intensity than the widely applied PAMcontaining dsDNA activator.The results presented in this study may help the fabrication of high-efficient CRISPRCas12a-based in vitro biosensing systems.
Key words:Cas12a;protospacer adjacent motif(PAM);dsDNA activator;ssDNA activator;in vitro biosensing
CRISPR(Clustered Regularly Interspaced Short Palindromic Repeats)and their associated protein(CRISPR-Cas)defend their prokaryotic hosts from invasion by foreign nucleic acids such as bacteriophage and plasmids[1-2].The CRISPR-Cas system uses programmable RNA to guide the Cas effector to the sequencespecific target and cleave the nucleic acid targets consequently.The well-studied Cas effector proteins,such as Cas9 and Cas12a(Cpfl),have been designed to be powerful gene editing tools[3-6].Unlike Cas9 systems,after a short T-rich protospacer-adjacent motif(PAM)[7]site recognition and CRISPR RNA(crRNA)binding to the target DNA,the structure of the Cas12a protein rearranges to form a catalytic activation conformation[8-10],activating the specificcis-cleavage activity along with thet rans-cleavage activity simultaneously[11-12],which can efficiently cut any single-stranded DNA(ssDNA)near the target DNA.Thetr ans-cleavage activity of the CRISPR-Cas12a system can significantly improve the sensitivity of nucleic acid detection in various applications,opening up a new channel forin vit ronucleic acid detection.Based on this,the researchers have established a range of CRISPR-Cas sensing platforms,such as DETECTR[12],HOLMES[13],and CONAN[14],etc.
More importantly,in addition to the direct recognition and detection of nucleic acid targets of interest or their replication products[15-17],thetrans-cleavage ability of Cas12a also allows for the indirect biosensing of various biomolecules by using pre-designed DNA activator sequences generated through certain targetresponsive behaviors,which greatly expands the application scope of the CRISPR technology.For example,Peng et al.use a modular catalytic hairpin assembly circuit to convert the quantitative information of microRNA into multiple programmable DNA duplexes that serve as the activators to initiate CRISPR-Cas12at ranscleavage activity[18].Li et al.describes an isothermal proximity CRISPR-Cas12a assay for protein detection where a pre-designed CRISPR-targetable sequence is achieved through proximity binding and extension to trigger Cas12a cleavage for signal amplification[19].Due to its great promise for fabricatingin vitrobiosensing assays,the fine-tuning oftrans-cleavage activity of the CRISPR-Cas system is significant to the detection of various types of biomolecules[20-22].
However,both dsDNA and ssDNA can be used as the activators to trigger thetr ans-cleavage activity of the CRISPR-Cas12a system,and the activator sequences used in the literatures are diverse and lack criterion for comparison.Therefore,how to systematically study the effect of DNA activator structures on thetrans-cleavage activity of Cas12a and find out the key influence factors will be of great significance for Cas12a-associated sensing system design and improvement of the detection sensitivity,which has not been comprehensively studied yet.In this contribution,we systematically designed DNA activators of different structures to regulate the binding process of Cas12a-crRNA-DNA activator and analyzed the effect of DNA activator structure on thetrans-cleavage activity of LbaCas12a(Fig.1).We find that there are some common rules for the structure design of dsDNA and ssDNA activators to trigger highertrans-cleavage activity of LbaCas12a,such as choosing canonical PAM sequences,reserving PAM-proximal overhang and truncating PAM-distal end sequence,etc.Based on these rules,a final-chosen DNA activator(rRNA)structure is recommended,which could generate a 3.7-time stronger fluorescence response than that yielded by the most widely used PAM-containing dsDNA activator.The relevant results are of great potential for the fabrication of high-sensitive CRISPR-Cas12aassociatedin vit robiosensing systems.
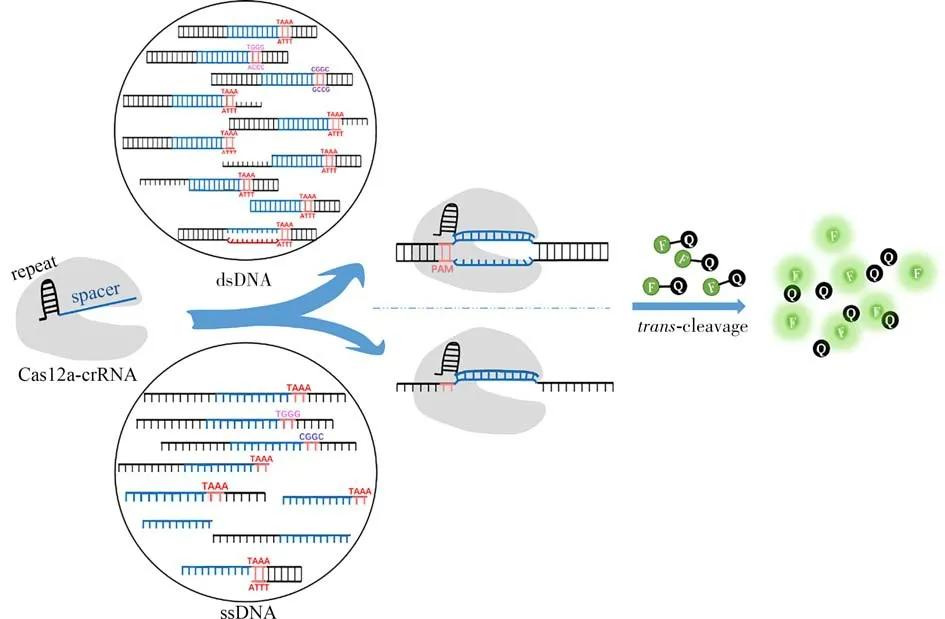
Fig.1 Schematic illustration of the principle used in this work
1 Experimental
1.1 Chemicals and materials
All oligonucleotides were synthesized by Thermo Fisher Scientific,and the sequence information is provided in Table 1.The underlined region in the crRNA is the spacer region.The PAM site in the DNA activators is highlighted in bold,and the sequences hybridized with the crRNA are underlined:

Table 1 Sequences of the crRNA and DNA activators
dsDNA-1(denoted as the standard DNA activator for comparison in this work)is formed by hybridization of strands TS-1 and NTS-1.dsDNA-2 is formed by hybridization of strands TS-2 and NTS-2.dsDNA-3 is formed by hybridization of strands TS-3 and NTS-3.dsDNA-4 is formed by hybridization of strands TS-4 and NTS-1.dsDNA-5 is formed by hybridization of strands TS-1 and NTS-4.dsDNA-6 is formed by hybridization of strands TS-4 and NTS-4.dsDNA-7 is formed by hybridization of strands TS-5 and NTS-1.dsDNA-8 is formed by hybridization of strands TS-1 and NTS-5.dsDNA-9 is formed by hybridization of strands TS-5 and NTS-5.dsDNA-10 is formed by hybridization of strands TS-1 and NTS-6.rDNA is formed by hybridization of strands TS-5 and NTS-7.
LbaCas12a(Cpf1)and 10×NEBuffer 2.1 were purchased from New England BioLabs(MA,U.S.A.).20×TE buffer(RNase-free,pH 7.5)and nuclease-free water were supplied by Thermo Fisher Scienti f ic.FQ reporter(5'-FAM-TTATT-BHQ1-3')was synthesized by Takara Biotechnology(Dalian,China).All the oligonucleotide solutions and buffers were prepared by nuclease-free water.
1.2 Preparation of DNA activators of different structures
The concentrations of all the oligonucleotides were quantitatively determined by Nanodrop 2000 and further diluted in nuclease-free water to a concentration of 2μmol/L.The dsDNA activator hybridization was conducted in a 10μL of reaction mixture containing 1μL TS strand,1.2μL NTS strand,and 7.8μL 1×NEBuffer 2.1.To generate dsDNA activator,the TS strand and NTS strand were annealed by incubation at 90℃for 3 min followed by slowly cooling to room temperature.The ssDNA activators were directly diluted in nuclease-free water to a concentration of 2μmol/L.
1.3 Evaluating LbaCas12a trans-cleavage activity by monitoring the fluorescence response
LbaCas12a and crRNA were mixed with a 1∶1 ratio(200 nmol/L∶200 nmol/L)in 1×NEBuffer 2.1 and pre-incubated at 37℃for 10 min to promote the complex formation.1μL DNA activator(final concentration of 20 nmol/L)and 1μL FQ reporters(final concentration of 1μmol/L)were then incubated with the mixture containing 1×NEBuffer 2.1 and 1μL LbaCas12a-crRNA(final concentration of 20 nmol/L)in a f inal volume of 10μL at 37℃for 50 min.The reactions were terminated by heating at 90℃for 3 min.Then,90μL of 1×TE buffer(20 mmol/L Tris-HCl,1 mmol/L EDTA,pH 7.9)was added to the reaction system,and the fluorescence signal was monitored at 520 nm on an F-7000 f luorescence spectrophotometer(Hitachi,Japan)with an excitation wavelength of 488 nm.
2 Results and discussion
2.1 crRNA and DNA activator designs
To ensure that the research results can faithfully reflect the influence of the activator structure but not other factors,three fundamental principles for the DNA activator and crRNA sequence design are established.First,the DNA activator itself does not have much non-specific hybridization,maintaining the stability of DNA double-stranded or single-stranded conformation.Second,except for the T-rich PAM site,there are no more than two consecutive T bases in the rest part of the DNA activator sequence to avoid non-specific recognition and cleavage.Third,make sure there are more than 17 complementary base pairs between the DNA activator and the spacer region of crRNA,which could facilitate the formation of Cas12a-crRNA/DNA activator ternary complex[23].The nucleotide chains used in this study all comply with the above principles to avoid representation and ensure the binding efficiency of Cas12a-crRNA complex and DNA activators not to be affected by the sequences.
Following the above principles,the effect of DNA activator structures on thetrans-cleavage activity of LbaCas12a system are systematically interrogated.Specifically,as illustrated in Fig.1,the Cas12a-crRNA complex combines with the activators of different structures to activate itstr ans-cleavage activity,cleaving the FQ reporters and generating a fluorescent signal.In this study,the strength of the fluorescent signal in the system was used to evaluate the activatedt rans-cleavage activity.
2.2 LbaCas12a trans-cleavage activity triggered by dsDNA activators
The dsDNA activators are the most commonly used DNA activation structures,and the combination of Cas12a-crRNA and dsDNA generally requires PAM sequence in dsDNA activators.Normally,Cas12a-crRNA recognizes PAM first,then the dsDNA activators unwind in the vicinity of the PAM,and the crRNA spacer sequence hybridizing with the TS strand to initiate R-loop formation and activate specificcis-cleavage activity,activating thetr ans-cleavage activity simultaneously[24].
First,we studied the influence of the PAM sequence on the dsDNA-induced LbaCas12atrans-cleavage activity.We selected TTTA,GCCG,and CCCA as potential PAM site next to the spacer-binding sequence and interrogated the LbaCas12atrans-cleavage ability activated by these three PAM-containing dsDNA activators(dsDNA-1,2,3,Fig.2).As displayed in Fig.3,the fluorescence signal of dsDNA-1 containing TTTA,the standard dsDNA structure,was higher than that of those induced by dsDNA containing either CCCA or GCCG.This indicates that LbaCas12a has a sequence preference for PAM recognition and a higher recognition efficiency toward the T-rich PAM sequence.This also demonstrates that LbaCas12a also recognizes the non-canonical C-rich PAM sequences(GCCG and CCCA),albeit of lower efficiencies than those with the canonical TTTA PAM[7,25].Therefore,the T-rich PAM sequence in the dsDNA activator is selected for subsequent research to activate the highertrans-cleavage activity of LbaCas12a.
Then,we explored whether the sequences adjacent to the PAM would have an impact on dsDNA activator recognition.By deliberately deleting the sequence fragment in the PAM-proximal end(the opposite of target site),we obtained three modified dsDNA activators(dsDNA-4,5,6,Fig.2)with certain deletions in single“target strand”(TS strand),single“non-target strand”(NTS strand)and double strands respectively.Compared to the standard dsDNA activator(dsDNA-1),the fluorescence signal gradually weakens(dsDNA-1>dsDNA-4~dsDNA-5>dsDNA-6,Fig.3).This indicates that the PAM proximal sequence has a certain influence on thet rans-cleavage activity of LbaCas12a.In the CRISPR-Cas12a systems,the crRNA guides LbaCas12a searching the PAM site to initiate the dsDNA activator recognition.Lacking PAM-proximal overhang may reduce the stability of the PAM site,thereby destroying the recognition and interaction of the PAM site by LbaCas12a.Taken together,the existence of the intact PAM-proximal overhang contributes to dsDNA positioning and enhancement of LbaCas12atrans-cleavage activity.
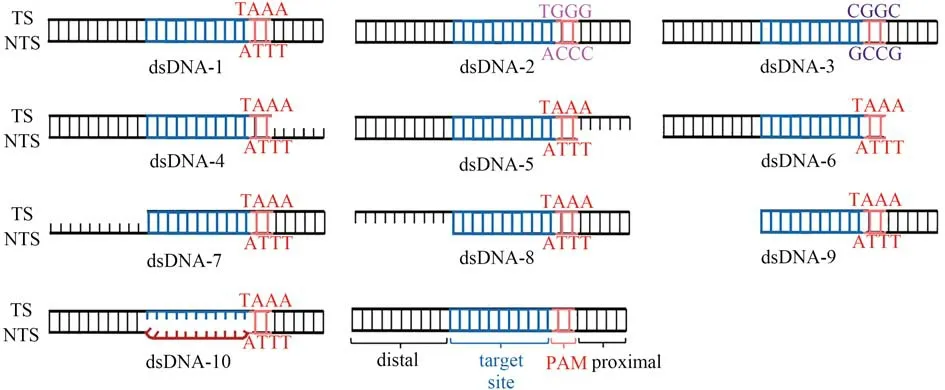
Fig.2 The structures of dsDNA activators
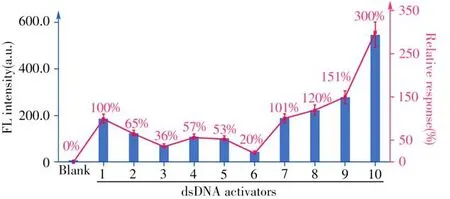
Fig.3 The fluorescence signals produced by different dsDNA activators(1-10),and the relative response compared to the PAM-containing dsDNA-1 as the standard(the fluorescence response of dsDNA-1 is normalized as 100%and the relative responses of other structures are provided in the brackets)the error bars ref lect the standard deviation of three parallel measurements
For the LbaCas12a system,after PAM site recognition,the unwinding of the strand in the vicinity of the PAM allows the TS strand to hybridize with the crRNA spacer sequence and then form an“R-loop”structure that activates the LbaCas12a conformational transformation,sequentially triggeringcis-cleavage of the NTS and TS strands.Thecis-cleavage product is combined with the Cas12a-crRNA complex,maintaining LbaCas12atrans-active state[23].We speculated that reducing the energy consumption of LbaCas12aci s-cleavage could contribute to the enhancement of itstr ans-cleavage ability.To verify this,we cut off the TS strand,NTS strand,and both strands of the PAM distal sequence,respectively(dsDNA-7,8,9,Fig.2)for studying the effect of the sequences at the PAM-distal end(outside of the target site)on LbaCas12at rans-cleavage activity.As displayed in Fig.3,the signal generated by dsDNA activator without PAM-distal end sequence(dsDNA-9)was 1.5 times higher than that induced by the standard dsDNA activator(dsDNA-1).Moreover,the NTS strand cancelling the PAM-distal sequence provides a higher enhancement factor on the LbaCas12at ranscleavage activity than the one with TS strand cancelled(dsDNA-8>dsDNA-7,Fig.3).We suppose that reducing the energy consumption of LbaCas12acis-cleavage will increase the fluorescence signal produced by LbaCas12atrans-cleavage in two aspects.On the one hand,reduction of the steric hindrance improves the flexibility of LbaCas12a conformational transitions.On the other hand,the newly activated LbaCas12a has a relatively high activity,which reduces the energy consumption ofcis-cleavage,and will retain more active LbaCas12a fortr ans-cleavage.
The R-loop is formed by directional crRNA-target hybridization.Previous research has demonstrated that R-loop formation is a key step in triggering the conformational transformation of LbaCas12a and activating itsciscleavage activity[10].We considered whether the formation of R-loop structure is related to thetrans-cleavage activity of LbaCas12a.By replacing the complementary NTS sequence with random sequence,crRNA was envisioned to hybridize with TS strand directly,leaving out the step of NTS displacement.Just as we expected,the fluorescence signal produced by dsDNA activator with pre-unwound structure(dsDNA-10)was significantly higher than that generated by fully hybridized TS/NTS strand(dsDNA-1).This result indicated that introducing unpaired protospacer into the dsDNA activator can accelerate the formation of R-loop,allowing a fast transition to ternary complex.Notably,the dsDNA activator with pre-unwound structure has generated the highest signal among all dsDNA activators,indicating that R-loop formation calls for most consumption of energy.
2.3 LbaCas12a trans-cleavage activity triggered by ssDNA activators
Besides regarded as the most widely investigated dsDNA,ssDNA activators have also been widely employed to activate thetrans-cleavage activity of LbaCas12a and fabricate efficient biosensing assays.
First,we verified whether the PAM sequence in ssDNA activators has the same effect on LbaCas12atranscleavage activity as dsDNA activators did.Here,we only used TS strands in the dsDNA activators(dsDNA-1,2,3,Fig.2)as ssDNA activators(ssDNA-1,2,3,Fig.4),and observed that the ssDNA activator with TAAA in the vicinity of the target site generated the strongest fluorescence signal(TAAA>CGGC>TGGG,Fig.5).This result demonstrated that partial PAM sequence in the ssDNA activator can also promote LbaCas12atrans-cleavage activity.
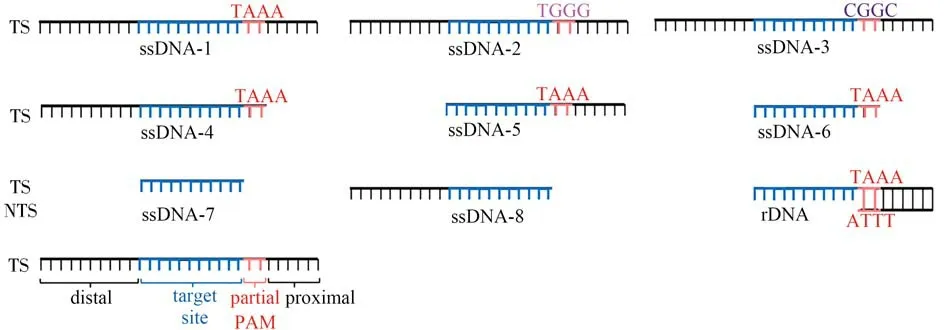
Fig.4 The structures of the ssDNA activators and the recommended optimal DNA activator(rDNA)
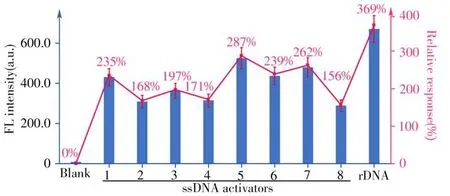
Fig.5 The fluorescence signals produced by different ssDNA activators(1-8)and the rDNA activator,and the relative response compared to the PAM-containing dsDNA-1 as the standard(normalized as 100%and the relative responses of other structures are provided in the brackets)
Then,considering the role of the PAM-proximal tail in positioning the whole chain,we removed the sequence fragment near to PAM from the ssDNA-1 and investigated the signal change(ssDNA-4,Fig.4).As expected,the ssDNA-4 activator produced 0.73-time of the fluorescence signal of the standard ssDNA-1 activator(Fig.5),proving that the ssDNA-4 activator has less chance of colliding with Cas12a-crRNA than ssDNA-1,which is similar to the results for dsDNA activators.This result reveals that when Cas12a-crRNA hunts the activator,the PAM-proximal tail will drive both the ssDNA activator and the dsDNA activator to easily contact with the Cas12a-crRNA,activating LbaCas12atrans-cleavage activity more efficiently.
Finally,we also tested whether the process that reducing energy consumption of thecis-cleavage can also apply to the ssDNA activators to increase LbaCas12atrans-cleavage activity.We cut off the sequence fragments adjacent to the crRNA-pairing sequence in the PAM-distal end of the ssDNA-1 and ssDNA-4,which were marked as ssDNA-5 and ssDNA-6(Fig.4).As shown in Fig.5,we can see that ssDNA-5 and ssDNA-6 activators without PAM distal sequence produced higher signals.Moreover,we extended the research object to ssDNA without PAM(ssDNA-7,8,Fig.4 and Fig.5),in which the similar results can be obtained.This further confirms our assumption.
2.4 General comparison of dsDNA and ssDNA activators
Throughout a comprehensive comparison,we can see that generally,ssDNA activators produce much higher signals than dsDNA activators(Fig.3 and Fig.5).By further combining with the finding that dsDNA activator with pre-unwound structure generates the highest signal output among all the dsDNA activators,we confirmed that ssDNA activators that do not need the formation of R-loop are better choices fori n vitrobiosensing.Based on the above findings,we proposed the optimal DNA activator design principle for the CRISPR-Cas12a system.(1)Using canonical PAM.(2)Preserving the tail at the PAM-proximal end.(3)Predeletion the fragment adjacent to the crRNA-pairing sequence at the PAM-distal end.(4)Omitting DNA unzipping.According to these design principles,we finally recommended a DNA activator(rDNA,Fig.4)structure that meets all the points mentioned above.The recommended rDNA shows the best performance,which produces 3.7 times higher signal than the standard dsDNA activator could(dsDNA-1,Fig.5).
3 Conclusions
In summary,we have systematically discussed the effect of DNA activators of different structures on the activation of CRISPR-Cas12atrans-cleavage activity towards potential applications fori n vi trobiosensing.It is found that thetr ans-cleavage activity of LbaCas12a is highly dependent on several critical steps,such as PAM recognition,activator positioning,R-loop formation,andci s-cleavage site trimming.As for the activator,no matter dsDNA or ssDNA,choosing the canonical PAM sequence,preserving the tail at the PAM-proximal end,and cutting off the PAM distal sequence are beneficial to enhance LbaCas12atrans-cleavage activity.Based on these interesting discoveries,a preferred DNA activator(rDNA)structure is recommended for fabricating biosensing systems,which can generate~3.7 times stronger signal than the standard dsDNA activator(dsDNA-1)does.This work not only offers more choices for DNA activator of Cas12a-associatedin vit robiosensing but also provides useful guidance for the structure design of DNA activators to enhance the detection sensitivity.

- 分析测试学报的其它文章
- Research Progress of Hemicyanine Dye for Molecular Imaging
- 碱性磷酸酶的体外检测和体内成像研究进展
- Recent Progress in Nanoscale MOFs for Biological Imaging of Tumors and Tumor Markers
- I-Motif-based Nanosystems for Biomedical Applications:p H Imaging,Drugs Controlled Release and Tumor Theranostics
- A Low-cost,Automated Nucleic Acid Extraction System Converted from the Open-Source Rep Rap 3D Printer
- Research Progress on Analytical Methods for Deciphering Adenosine-to-inosine RNA Editing