
Research Progress on Analytical Methods for Deciphering Adenosine-to-inosine RNA Editing
2022-04-22 03:36CHENJuanjuanYUANBifengFENGYuqi
分析测试学报 2022年4期
CHEN Juan-juan,YUAN Bi-feng,2*,FENG Yu-qi,2
(1.Sauvage Center for Molecular Sciences,Department of Chemistry,Wuhan University,Wuhan 430072,China;2.School of Public Health,Wuhan University,Wuhan 430071,China)
Abstract:Adenosine-to-inosine(Ade-to-Ino)RNA editing is one of the most widespread post-transcriptional modifications in RNA.The conversion of adenosine to inosine is through the hydrolytic deamination of the amino group at the C6 position of adenosine,which is catalyzed by the adenosine deaminases acting on RNA(ADARs).Accumulated lines of evidence show that Ade-to-Ino RNA editing is involved in the regulation of gene expression and protein functions.Aberrant Ade-to-Ino RNA editing is demonstrated to be correlated with many human diseases.Indepth investigation of the biological functions of Ade-to-Ino RNA editing depends on the sensitive detection,accurate quantification,and precise mapping analysis.This review provides an overview of the recent advances in analytical methods and techniques for the detection,quantification and location analysis of Ade-to-Ino RNA editing in a wide range of RNA molecules.The principles,advantages,limitations and applications of these established methods are discussed.It is hoped that this review could stimulate the development of analytical methods to decipher Ade-to-Ino RNA editing and expedite the elucidation of the functions of Ade-to-Ino RNA editing in various RNA species.
Key words:adenosine-to-inosine RNA editing;RNA modification;detection method;mapping analysis
DNA and RNA molecules contain various modifications that play critical roles in a wide variety of biological processes[1-6].Over 150 different types of modifications have been identified to be present in different RNA species of living organisms[7-9].Adenosine-to-inosine(Ade-to-Ino)RNA editing is one of the most widespread post-transcriptional modifications in RNA of mammals[10].In Ade-to-Ino RNA editing,adenosine in RNA is deaminated to form inosine through hydrolytic deamination of the amino group at the C6 position of adenosine,by enzymes called adenosine deaminase acting on RNA(ADAR)family proteins(Fig.1A)[11].Three ADAR proteins have been identified in mammals.Enzymatically active ADAR1 and ADAR2 are typically expressed in most mammalian tissues,while ADAR3 that lacks enzymatic activity is exclusively expressed in the brain of mammals[11].Ade-to-Ino RNA editing can change RNA sequences,coding potential and secondary structure,but doesn′t alter the sequences of genomic DNA.

Fig.1 Schematic illustration of the formation of Ino and its base pair with cytosine
A plenty of Ade-to-Ino RNA editing events have been identified in various types of RNA,including messenger RNA(mRNA)[12],transfer RNA(tRNA)[13],microRNA(miRNA)[14]and long non-coding RNA(lncRNA)[15].Ino prefers to base-pair with cytosine(C)and is therefore recognized as guanosine(G)by the translation machinery(Fig.1B).As a result,Ade-to-Ino RNA editing can change the genetic information after transcription.Ade-to-Ino editing occurring in the coding region of mRNA can induce amino acid substitutions in translated proteins,leading to the increased diversity of proteins.Ade-to-Ino editing in the non-coding RNA can modulate the stability,localization,and splicing of RNA[16-17].
Ade-to-Ino RNA editing has important functions and biological significance.A growing number of studies have shown that Ade-to-Ino RNA editing is involved in the regulation of gene expression and protein functions,and can modulate many biological processes.Aberrant Ade-to-Ino RNA editing has been found to be correlated with many human diseases,including a variety of cancers(head and neck squamous cell carcinoma,HNSC;glioblastoma multiforme,GBM;thyroid carcinoma,THCA;breast invasive carcinoma,BRCA;lung adenocarcinoma,LUAD;liver hepatocellular carcinoma, LIHC; kidney renal papillary cell carcinoma,KIRP;kidney chromophobe,KICH;kidney renal clear cell carcinoma,KIRC;colon adenocarcinoma,COAD;bladder urothelial car cinoma,BLCA)[18-21],neurological and neurodegenerative diseases[22],psychiatric disorders[23]and autoimmune diseases[24](Fig.2).Ade-to-Ino RNA editing has been shown to contribute to disease pathologies with the editing occurring in glutamate receptors,serotonin receptors,and gamma-aminobutyric acid receptors,etc[25-26]. These findings highlight the dysregulation of Ade-to-Ino RNA editing in the pathogenesis of human diseases.
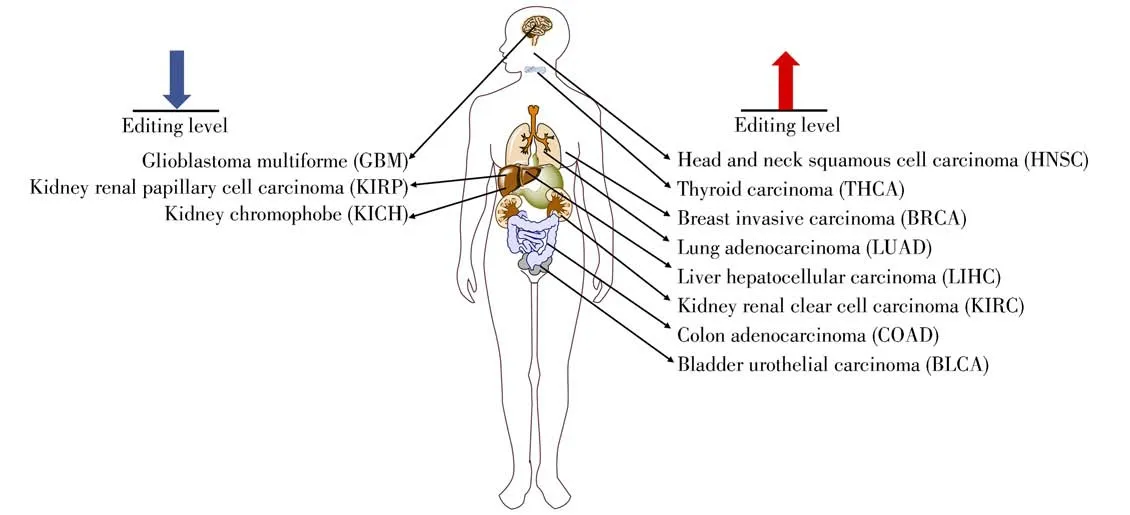
Fig.2 Aberrant Ade-to-Ino RNA editing level correlated with various human diseases
In-depth investigation of the biological functions of Ade-to-Ino RNA editing depends on the sensitive detection,accurate quantification,and precise mapping analysis.Here,we review and categorize the methods and techniques for detecting Ade-to-Ino RNA editing,including the overall detection,site-specific detection,and transcriptome-wide mapping analysis(Fig.3).We discuss the principles,advantages,limitations and applications of these established methods.
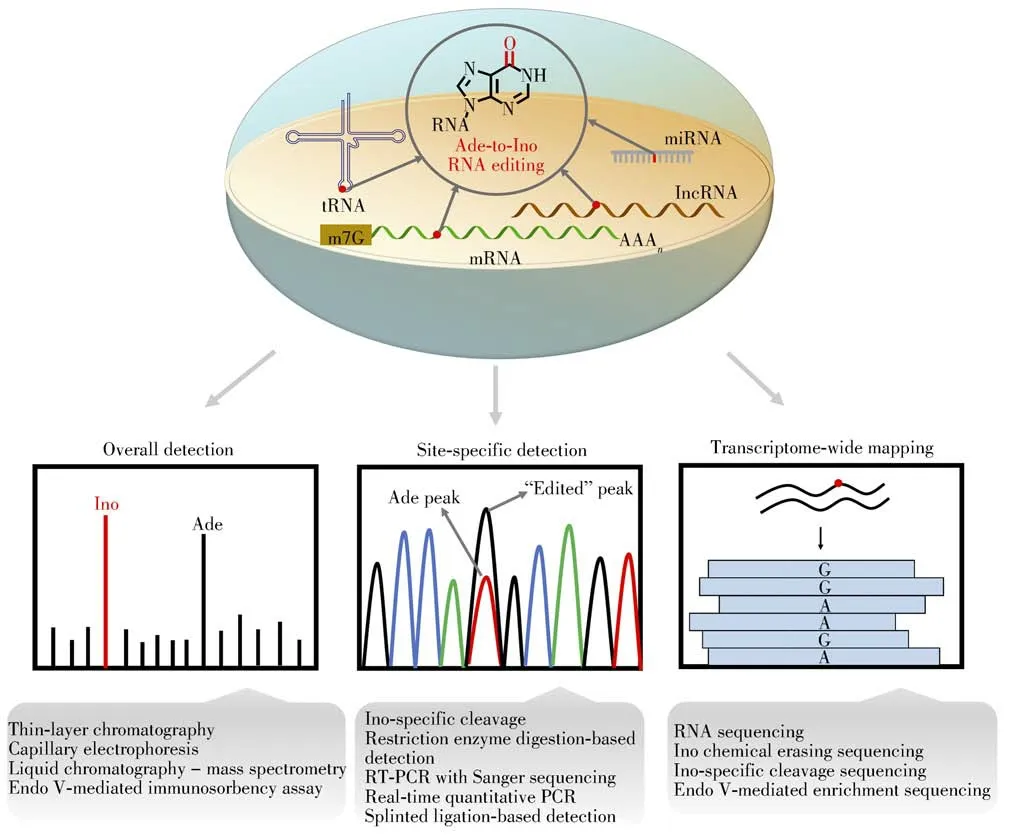
Fig.3 Schematic illustration for the overall detection,site-specific detection,and transcriptome-wide mapping analysis of Ade-to-Ino RNA editing
1 Methods for overall detection of Ade-to-Ino RNA editing
The established methods for overall detection of Ade-to-Ino RNA editing mainly include thin layer chromatography(TLC),capillary electrophoresis(CE),liquid chromatography-mass spectrometry(LCMS),and Endonuclease V(Endo V)-mediated immunosorbency assay(Table 1).However,these methods generally cannot provide the positional information of Ino.

Table 1 Limit of detection(LOD),linear range,calibration curve and sample input for the overall detection of Ino
1.1 Thin layer chromatography
Thin layer chromatography(TLC)followed by autoradiography or UV spectrophotometric was developed to detect Ino in mRNA in 1998[27].As for TLC-based detection,mRNA was enzymatically digested to monophosphates(NMPs)by nuclease P1,followed by radiolabeling with P32or UV detection.With this method,the measured level of Ino(Ino/all nucleotides)in mRNA from different rat tissues was in the range of 0.000 7%~0.006%[27].2D(two dimension)-TLC could detect and quantify RNA modifications down to femtomole level including Ino[33].However,the TLC analytical procedure is relatively time-consuming and involved in radioactive procedures that required additional certificate to be carried out in laboratory.
1.2 Capillary electrophoresis
The principle of capillary electrophoresis(CE)is based on the separation of charged particles in an electric field[34].Nucleotides are negatively charged in the pH 2-12,therefore they can be readily separated by CE[35].
Cornelius et al.[28]applied capillary electrophoresis with laser-induced fluorescence(CE-LIF)to detect Ino based on the detection of ribonucleoside-5'-monophosphates that were conjugated with 4,4-difluoro-5,7-dimethyl-4-bora-3a,4a-diaza-s-indacene-3-propionyl ethylene diamine hydrochloride(BODIPY FL EDA)at the 5'-phosphate group.After enzymatic digestion of RNA to 5'-monophosphates by nuclease P1 and BODIPY labeling,the BODIPY conjugates were determined by CE-LIF without further purification.The limit of detection(LOD)for Ino by this method was 160 pmol/L.Recently,2,4,6-trinitrobenzenesulfonic acid was employed to label Ino to form fluorescent trinitrophenylated complexes of TNP-Ino,which exhibited~25-fold fluorescence enhancement after the formation of inclusion complexes withγ-cyclodextrin[29].With this method,Ino in whole tissue homogenates of rat forebrain was quantified with the measured level of Ino being(6.4±0.8)nmol/mg.
1.3 Liquid chromatography-mass spectrometry
Mass spectrometry(MS)has been widely used to detect and quantify nucleic acid modifications as MS exhibits high detection sensitivity and good capability to identify compounds[36-39].LC-MS is a widely used platform for the qualitative and quantitative detection of RNA modifications[40-44].
Our group has developed LC-MS method for analyzing a variety of nucleic acid modifications[45-51].As for the detection of Ino in RNA,RNA samples are first enzymatically digested to nucleosides,which are then extracted by chloroform to remove the nucleases and finally analyzed by LC-MS with multiple reaction monitoring(MRM)detection mode[12].With this method,the measured level of Ino(Ino/Ade)in mRNA from HEK293T cells was 0.008%±0.000 3%[12].We further found that Cr(Ⅵ)exposure could induce an obvious decrease of Ino in mRNA,indicating that Cr(Ⅵ)could interrupt Ade-to-Ino RNA editing in mRNA.With the established LCMS method,we also quantified Ino in mRNA and small RNA(<200 nt)from thyroid carcinoma and matched tumor-adjacent normal tissues[30].We found that Ino in small RNA(<200 nt)exhibited significant increase in thyroid carcinoma tissues compared to the normal tissues.Grobe et al.[31]detected and quantified RNA modifications in total tRNA fromPseudomonas aeruginosaPA14,with the measured level of Ino(Ino/all ribonucleosides)being 0.003 26%±0.001 76%.LC-MS-based detection method is highly quantitative and non-radioactive.However,this method is relatively low throughput and necessitates specialized equipment.
1.4 Endo V-mediated immunosorbency assay
Endo V,a conserved nucleic acid repair enzyme,can specifically recognize Ino in nucleic acids[52-54].Endo V utilizes Mg2+to cleave Ino-containing substrates at the second phosphodiester bond 3'to Ino.The enzyme activity can be modulated by replacing Mg2+with Ca2+,enabling Endo V to bind instead of to cleave Inocontaining nucleic acid substrates[55].With this unique property,Endo V linked immunosorbency assay(Endo VLISA)was proposed to measure the global Ade-to-Ino editing level in cellular RNA[32].In this work,terminal 3′OH of cellular RNA was first oxidized to generate 3′dialdehyde by NaIO4,followed by denaturing RNA with glyoxal.RNA 3'terminal was biotinylated by the condensation of 3′dialdehyde and biotin-PEG4-hydrazide,enabling to immobilize RNA onto streptavidin-coated wells.Then the wells were probed with commercial eEndo V in the presence of Ca2+,and Ino-RNA specifically bound with commercial eEndo V[56].Since the commercial eEndo V was fused to a maltose-binding protein(MBP)affinity tag,the wells were then probed with a mouse anti-MBP primary antibody and goat anti-mouse secondary antibody conjugated to horseradish peroxidase to produce a chemiluminescent signal.With the Endo VLISA method,~100 fmol Ino per μg mRNA could be reliably detected.
2 Methods for site-specific detection of Ade-to-Ino RNA editing
The reported methods for site-specific detection of Ade-to-Ino RNA editing mainly include Ino-specific cleavage,restriction enzyme digestion-based detection,RT-PCR with Sanger sequencing,real-time quantitative PCR,and splinted ligation-based detection.The editing efficiencies of specific sites can be determined by site-specific detection.
2.1 Ino-specific cleavage
Ino-specific cleavage is performed by reacting RNA with glyoxal,stabilizing the glyoxal adducts with borate,and enzymatically digesting RNA with ribonuclease T1(RNase T1).RNase T1 cleaves RNA strand after G or Ino.Glyoxal can form a stable adduct with G but not with Ino[57].The glyoxal-protected G is resistant to RNase T1 cleavage.Thus,glyoxalated RNA is cleaved only after Ino by RNase T1[58],which can be employed to determine Ino site in RNA.
A selective amplification method with Ino-specific cleavage was proposed to identify Ade-to-Ino RNA editing sites[58].In this work,mRNA was first treated with glyoxal and borate to protect G,followed by RNase T1 digestion.After glyoxal removal and anchor ligation,extension of first-strand cDNA was performed by reverse transcription using an anchor primer.Ino-containing fragments were preferentially amplified using sequence specific upstream primers and discriminating anchor primers(DAPs)for the downstream primer.The DAPs were designed to end with three extra nucleotides of the form“CNX”,where the C paired with the G in cDNA that originated from Ino.With this method,the glutamine to arginine(Q/R)and arginine to glycine(R/G)editing sites on GluR-B mRNA in the rat brain were identified.Based on Ino-specific cleavage with exon array analysis,specific Ade-to-Ino edited transcripts were also determined,such as Gria2,Htr2c,Gabra3 and Cyfip2 in mouse brain[59].
2.2 Restriction enzyme digestion-based detection
Restriction enzyme digestion-based method was developed to detect Q/R editing site of GluR2 mRNA[60].In this method,RT-PCR was used to amplify a region across the Q/R editing site of GluR2 mRNA.The restriction enzyme ofBbvIrecognizes the sequence 5'-GCAGC-3'andAciIrecognizes the sequence 5'-GCGG-3'.Therefore,BbvIcleaves the PCR products at Q/R site originating from unedited mRNA,andAciIcleaves the PCR products at Q/R site deriving from edited mRNA.The subsequent electrophoresis analysis of the restriction enzyme-digested products enabled the quantification of the Ade-to-Ino editing level.With this method,it was found that Q/R site was nearly 100%edited on GluR2 mRNA in the epileptic hippocampus and temporal cortex.The Q/R editing efficiency was significantly increased on GluR5 and GluR6 mRNA in the epileptic temporal cortex[60].Editing of R/G site on GluR2 mRNA was elevated in the epileptic hippocampus[61].The Q/R editing efficiency on GluR2 mRNA was significantly reduced in the spinal ventral gray of amyotrophic lateral sclerosis[62].However,this method was limited to the analysis of Ade-to-Ino editing in the recognized sites by restriction enzymes.
2.3 RT-PCR with Sanger sequencing
RT-PCR combined with Sanger sequencing was established to identify and quantify Ade-to-Ino editing at specific sites of interest.The reverse transcribed products were amplified by PCR followed by Sanger sequencing.Overlapping peaks of A and G could be observed for the partially edited sites.The peak areas of A and G could be utilized to estimate the Ade-to-Ino RNA editing level[63-64].With this method,it was observed that the editing level of isoleucine to valine(I/V)site on the potassium channel Kv1.1 mRNA was downregulated during the course of nonfamilial temporal lobe epilepsy[65].In Alzheimer′s hippocampus,an average 4%decrease of Ade-to-Ino editing level of GluA2 mRNA at Q/R site was confirmed[66].
Direct Sanger sequencing is not capable to determine the Ade-to-Ino RNA editing if the editing level is low.In this respect,colony sequencing has been employed to quantify Ade-to-Ino RNA editing.The Ade-to-Ino RNA editing level at specific site could be calculated as ratio of the clonal numbers of G over the sum numbers of A and G.With this method,5.1%of the Kv1.1 mRNA was edited at I/V site in entorhinal cortex of wild-type rats,whereas 21.5%of the Kv1.1 mRNA was edited at I/V site in entorhinal cortex of chronic epileptic rats[67].Compared to direct Sanger sequencing,the colony sequencing offers a more accurate quantification of the editing level at specific sites.
2.4 Real-time quantitative PCR
Chen et al.[68]developed real-time quantitative PCR(qPCR)using SYBR Green to quantify Ade-to-Ino editing at the Q/R site of zebrafishgri a2amRNA and tyrosine to cysteine(Y/C)site of zebrafishgrik 2amRNA.After RT-PCR,A originating from unedited mRNA and G originating from edited mRNA were quantified by the universal primer pair annealing to both,while A originating from unedited mRNA was quantified by 3′ends of A-specific primers placed at the editing site.With this method,it was found that 91.2%of thegria2amRNA was edited at Q/R site in the 24-hpf zebrafish embryo,while 44.6%of thegrik 2amRNA was edited at Y/C site in 24-hpf zebrafish embryo.
Real-time qPCR with using TaqMan probes was also developed to determine Ade-to-Ino RNA editing frequencies at specific sites[69].In this strategy,the region across the editing site was amplified by two PCR primers,and the edited mRNA and unedited mRNA were simultaneously detected by G(VIC)probe and A(FAM)probe,respectively.To measure the editing frequency of the Q/R site on GluR5 mRNA,each experimental cDNA sample and each dilution of the A and G standards as templates were amplified by multiplex qPCRs(containing two probes).The amount of edited or unedited cDNA of the experimental sample was calculated according to standard curve of the G or A,respectively.With this method,the mean editing efficiency of the Q/R site on human GluR5 mRNA was estimated to be 62.31%±6%.
2.5 Splinted ligation-based detection
Ino at position 34(Ino34)is present in many tRNAs,such as t RNAArgACGin bacteria and tRNAArgACG,tRNAAlaAGC,t RNAProAGG,tRNAThrAGU,tRNAValAAC,tRNASerAGA,tRNALeuAAG,and tRNAIleAAUin eukaryotes,while Ino is absent in archea[70].A reverse transcription-free method called“splinted ligation-based inosine detection”(SL-ID)was developed for detecting Ino in t RNA[71].In this work,total RNA was enzymatically cleaved byThermotoga marit i maEndo V(tmEndo V)to generate the Ino 34-containing t RNA half,which was subsequently captured by a specific DNA bridge that was also complementary to a 3′-32P-ligation DNA.The tRNA half and the ligation oligonucleotide were ligated by the T4 DNA ligase,followed by dephosphorylation of the remaining 3′-32P-ligation DNA.The internally labeled32P-ligation product was preserved.The subsequent denaturing polyacrylamide gel electrophoresis(PAGE)analysis of expected ligation products enabled the identification of the Ade-to-Ino editing.With this method,Ino34 on endogenous tRNAValAAC,tRNAArgACG,tRNAThrAGU,and tRNAAlaAGCfrom HeLa cells and on endogenous tRNAValAAC,tRNAArgACGfrom HEK293T cells were
[72-73]identified.Although splinted ligation could be quantified,SL-ID based on the tmEndo V was not suitable to quantify the editing level of Ino34 as it exhibited low efficiency for Ino34 cleavage.
3 Methods for transcriptome-wide mapping of Ade-to-Ino RNA editing
3.1 RNA sequencing
Methods for overall detection and site-specific detection have limitations for identifying Ade-to-Ino RNA editing in large-scale.The rapid advancement of the high-throughput sequencing enables the transcriptomewide mapping of Ade-to-Ino editing(Fig.4).
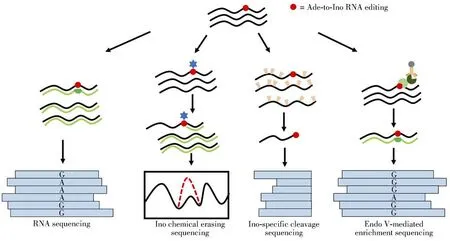
Fig.4 Summary of the analytical strategies for transcriptome-wide mapping of Ade-to-Ino RNA editing
Direct RNA sequencing(RNA-seq)is a straightforward method for identifying Ade-to-Ino editing sites in transcriptome.As Ino prefers to pair with C during reverse transcription,Ino is read as G in sequencing.Thus,the sites partially or completely replacing with G(A-to-G conversion)are candidates for Ade-to-Ino editing.However,RNA-seq may have high false positives due to the sequencing errors,alignment errors,single nucleotide polymorphisms(SNPs),somatic mutations,and spontaneous chemical alterations[74].Therefore,advanced bioinformatics methods to eliminate false positives,such as statistical modeling,filtering and/or alignment,and integration of additional genomic information were developed to improve the accuracy of the method[75].
RNA-seq has been applied to transcriptome-wide mapping of Ade-to-Ino editing in various cancer tissues and cells.Elevated Ade-to-Ino editing levels were detected in many tumor tissues,including breast,thyroid,head and neck,and lung cancers[20-21].On the contrary,Ade-to-Ino editing level in brain tumor tissues was generally downregulated[76].In addition,Ade-to-Ino editing of miRNAs was also characterized by RNA-seq.Wang at al.[77]characterized miRNA editing profiles of 20 cancer types and identified 19 Ade-to-Ino editing hotspots in miRNAs.As for neurological diseases,256 Ade-to-Ino editing sites in 87 genes were differentially edited between epileptic mice and healthy controls[78].So far,millions of Ade-to-Ino RNA editing sites from various organisms,tissues and genomic regions in human were identified and published online in different databases,such as REDIportal[79],RADAR[80],DARNED[81].Notably,databases related to diseases have also been established,such as ADeditome for Alzheimer′s disease[82]and GPEdit for cancers[83],which offers helpful information for investigating the functions of the Ade-to-Ino editing.
3.2 Ino chemical erasing sequencing
Ino can be cyanoethylated to formN1-cyanoethylinosine(ce1Ino)by acrylonitrile through Michael addition(Fig.5A)[84].TheN1-cyanoethyl group of ce1Ino inhibited Watson-Crick base-pairing with C,as a result,synthesis of a first-strand cDNA was arrested at ce1Ino during reverse transcription.Inosine chemical erasing(ICE)method was developed to identify Ade-to-Ino editing sites in the human transcriptome in 2010(Fig.5B)[85].In ICE method,Ino in RNA was cyanoethylated with the acrylonitrile treatment,while RNA without acrylonitrile treatment was prepared as a control(i).These RNAs were then subjected to first-strand cDNA synthesis by reverse transcription(ii).For partially edited sites,the unedited A and the edited Ino of untreated RNA are converted to thymidine(T)and C,respectively,in the cDNAs.In the cyanoethylated RNA,ce1I arrested extension of the cDNA at the editing site.These cDNAs were then amplified by PCR(iii),followed by direct sequencing(iv).In the control,overlapping peaks of A and G were displayed at the Ade-to-Ino editing sites in the sequencing chromatogram.However,in the cyanoethylated RNAs,the G peak that was substituted for the Ino disappeared,and an A peak was displayed at the editing sites.With the ICE method,5 072 Ade-to-Ino editing sites in the human transcriptome were identified.
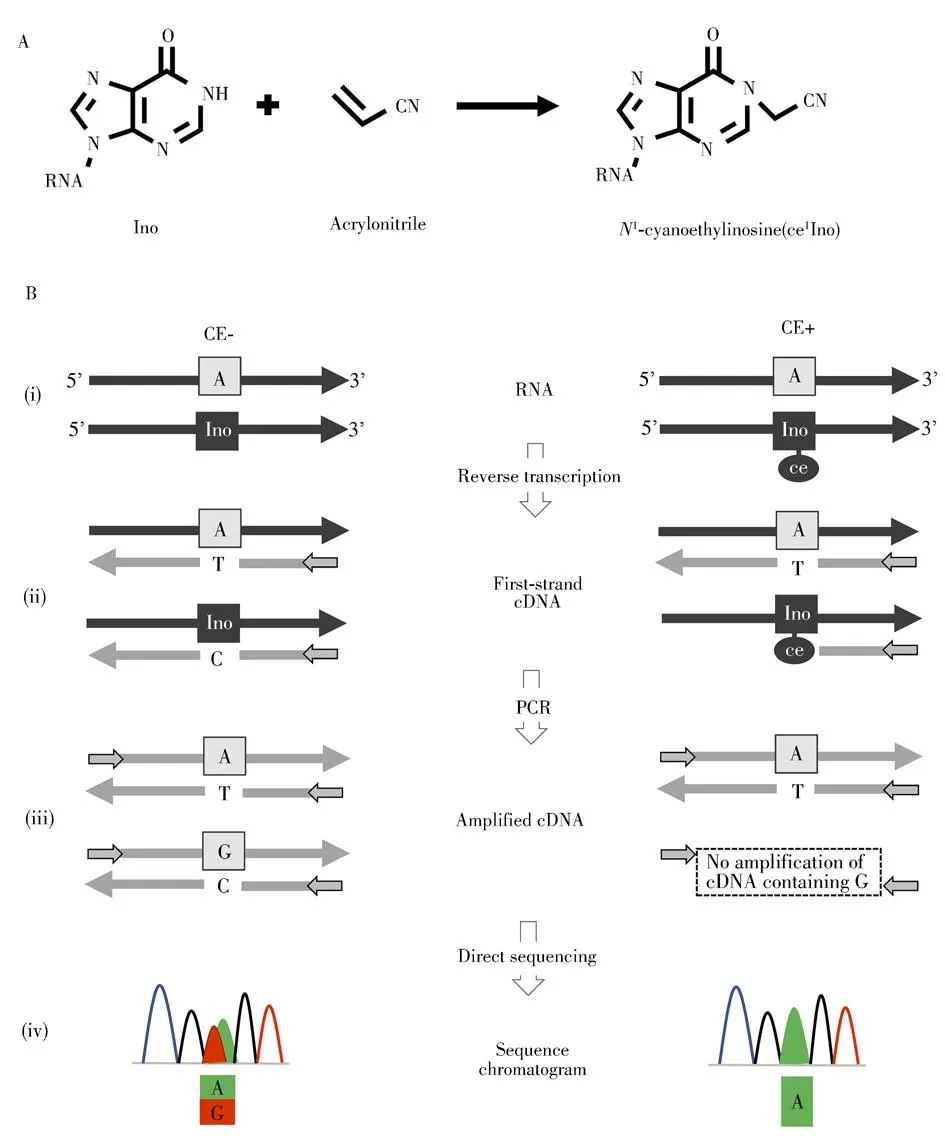
Fig.5 The principle and analytical procedure of ICE-seq method
For global and unbiased mapping of Ade-to-Ino editing in human transcriptome,Suzuki′group[86-87]combined ICE method with high-throughput sequencing to generate an efficient strategy called“ICE-seq”.With the ICE-seq,19 791 sites were identified and 1 258 edited mRNAs were found,including 66 sites in coding regions,41 of which could alter amino acid assignment.The ICE-seq method,however,is not capable of detecting Ino sites with 100%Ade-to-Ino editing.Moreover,if multiple Ino modifications locate in close range,some Ino sites may be lost.
3.3 Ino-specific cleavage sequencing
Cattenoz et al.[88]combined Ino-specific cleavage with high-throughput sequencing to map Ade-to-Ino RNA editing in the mouse transcriptome.In this method,RNA was first oxidized and biotinylated at 3′ends,followed by the glyoxal and borate treatment to protect G.Glyoxal RNA was then immobilized to streptavidin magnetic beads by biotin-streptavidin affinity coupling.After treatment with RNase T1,only Ino-RNA could be cleaved and removed from magnetic beads for library preparation and high-throughput sequencing.With this method,665 Ade-to-Ino editing sites were identified in mouse brain RNA.However,if G was not completely protected by glyoxal,it could be also cleaved by RNaseT1,resulting in false positive recognition of Ade-to-Ino editing.
3.4 Endo V-mediated enrichment sequencing
Endo V inosine precipitation enrichment sequencing(Endo VIPER-seq)method was developed for mapping Ade-to-Ino editing in human brain mRNA[56].In this work,eEndo V was utilized to recognize and capture Ino-RNA in the presence of Ca2+.Since the commercial recombinant eEndo V was fused to an MBP tag,the complex of Ino-RNA and eEndo V could be pulldown by anti-MBP functionalized beads.In Endo VIPERseq,cellular mRNA was first fragmented to~200-500 nt followed by glyoxal treatment to denature RNA.Inocontaining RNA was then subjected to eEndo V precipitation enrichment and the resulting RNA was used to library preparation and high-throughput sequencing.The Endo VIPER-seq method enabled the~1.8-fold higher identified number of Ade-to-Ino RNA editing sites than that by direct RNA-seq.Endo VIPER-seq overcame the limitation that notable amounts of initial RNA material and/or huge numbers of sequencing reads were needed to achieve enough depth and coverage in RNA-seq.
4 Conclusions
Since the discovery in 1965 in yeast tRNAAla[89],Ino has emerged as a widespread modification of RNA with diverse functions.The identification and detection of Ade-to-Ino RNA editing sites has been largely promoted by the development of advanced technologies.In this review,we summarize the methods developed for the detection,quantification,and location analysis of Ade-to-Ino RNA editing sites in a wide range of RNA molecules.Although LC-MS-based detection is the most widely used method for quantifying Ino,direct LCMS analysis is challenging when quantifying Ino in RNA species with extremely low abundance.In this respect,chemical labeling combined with LC-MS analysis will be a good choice to increase the detection sensitivity.The tagged group added to the Ino from labeling reagents can be utilized to improve ionization efficiency of Ino during MS analysis.As for transcriptome-wide mapping analysis of Ino,we expect that more straightforward bioinformatics strategies will be established for accurately identifying Ino in RNA.
The past decade has witnessed the fast advancement of the study of Ade-to-Ino RNA editing.However,there still are many important questions that haven′t been addressed.For examples,how is the process of Ade-to-Ino RNA editing regulated?What are the mechanisms used by ADAR enzymes to target specific adenosine in RNA?What are the functions of each Ade-to-Ino RNA editing site in RNA?Revealing the physiological roles of RNA editing in human diseases is still a challenging task.Nevertheless,with the advances in analytical technologies and data processing,we envision that the impact of Ino on human diseases will be better understood.

- 分析测试学报的其它文章
- Research Progress of Hemicyanine Dye for Molecular Imaging
- 碱性磷酸酶的体外检测和体内成像研究进展
- Recent Progress in Nanoscale MOFs for Biological Imaging of Tumors and Tumor Markers
- I-Motif-based Nanosystems for Biomedical Applications:p H Imaging,Drugs Controlled Release and Tumor Theranostics
- A Low-cost,Automated Nucleic Acid Extraction System Converted from the Open-Source Rep Rap 3D Printer
- CRISPR/Cas Based Biosensing Platforms for Molecular Diagnosis