
合金元素在fcc-Fe/NbX(X=C,N)界面的偏析及影响
2022-04-19 09:04姜周华韩培德
金属热处理 2022年4期
杨 静, 董 楠, 姜周华, 韩培德
(1. 太原理工大学 材料科学与工程学院, 山西 太原 030024; 2. 东北大学 冶金学院, 辽宁 沈阳 110004)
超级奥氏体不锈钢与普通奥氏体不锈钢相比,含有更高的Cr、Ni、Mo、N等元素,具有优异的抗局部腐蚀和应力腐蚀能力,广泛应用于烟气脱硫、海水淡化以及石油化工等领域[1-2]。随着使用环境的恶化,对材料的强度、耐蚀性、耐热性等不断提出新的要求,Mo含量达6%~7%的高钼超级奥氏体不锈钢在某些情况下可与具有极佳耐蚀性的C276等铁镍基合金相媲美。而其强度主要通过固溶强化来提高,但提高幅度有限[3]。析出强化是最有效的强化方式之一,通过弥散分布的析出相可显著提高不锈钢的屈服强度与抗拉强度,由于细小的析出相钉扎位错和晶界,抑制了热处理过程中的晶粒长大,晶粒细化有效地提高了钢的强韧性。
Nb、Ti在不锈钢中既可提高耐腐蚀性又可提高强度,奥氏体不锈钢中Ti、Nb与C或N结合形成细小、弥散的MX(NbC、TiC、NbN、TiN)沉淀相,对位错和其他晶格缺陷的钉扎起到更好的强韧化效果,通常将Nb-Mo复合加入微合金钢中实现元素的优势互补。研究发现相较于Nb-Ti微合金钢,Nb-Mo微合金钢中的碳化物更弥散、细小。Zhou等[4]利用EDS和HR-FEM技术,在304H钢中发现Nb(C, N)周围有σ相的存在,对Cr原子在奥氏体不锈钢中Nb(C, N)相界面的分布进行了分析,发现在界面处Cr有富集现象,其可能是σ相形成的前期。一些学者[5]将Nb加入超级奥氏体不锈钢并进行了研究,Nb可提高超级奥氏体不锈钢的耐蚀性能,但是形成的NbC与σ相之间有无关联性,尚未见相关报道。目前,针对不锈钢中添加Nb,分析NbC析出相,以及bcc-Fe/NbC、fcc-Fe/NbN界面特性的试验、理论研究已经很多[6-7]。但是围绕合金元素,尤其Mo在fcc-Fe/NbC或fcc-Fe/NbN界面偏析行为的研究还很少。Mo为易偏析元素,极易形成σ相,其若析出于fcc-Fe/NbC或fcc-Fe/NbN界面,对于超级奥氏体不锈钢是不利的[8-9]。因此很有必要开展相关研究,本文基于此,利用第一性原理研究合金元素Si、Ni、Mn、Cr、Mo等在fcc-Fe/NbX(X=C,N)界面的偏析行为,为高性能超级奥氏体不锈钢的合金化设计提供指导。
1 计算方法及模型建立
1.1 计算方法
本文所有计算均使用Vienna ab initio simulation package (VASP)软件进行,运用Perdew burke-ernzerhof (PBE)在半局部DFT级的广义梯度近似(GGA)中的交换-相关函数Projector augmented wave (PAW)方法。最大平面波截断能为400 eV,K点设置为8×8×1。自洽场迭代的力收敛阈值设置为10-5eV,作用于每个原子上的力不大于0.3 eV/nm,内应力不大于0.05 GPa。由于奥氏体不锈钢无磁性,且考虑计算速度,本文计算均不加自旋。NbX(X=C,N,下同)和fcc-Fe存在Baker-Nutting取向关系,故本研究搭建的是(001)fcc-Fe/(001)NbX界面模型。讨论了不同层数及终端的NbX界面,最终选取了5层NbX、9层fcc-Fe。
1.2 fcc-Fe/NbX界面结构模型
由NbX、fcc-Fe块体结构建立了5个Nb、5个C/N原子的5层NbX及18个Fe原子的9层fcc-Fe的fcc-Fe/NbX界面原子结构模型(共28个原子),见图1。真空层的厚度为1 nm,以避免周期性边界条件引起的相邻界面之间的相互作用。界面结构体系晶格参数为a=b=0.328 84 nm,c=3.544 30 nm,α=β=γ=90°。图1中编号1至5位置为计算中所替换原子的替换位点,替换原子M(Si、Ni、Mn、Cr、Mo)。为了保证界面模型中各原子数守恒,故替换原子位于1及2位点(取代Fe)时,3位点为Fe原子;替换原子位于4及5位点(取代Nb)时,3位点为Nb。替换原子位于3位点时,其余位点原子保持不变。
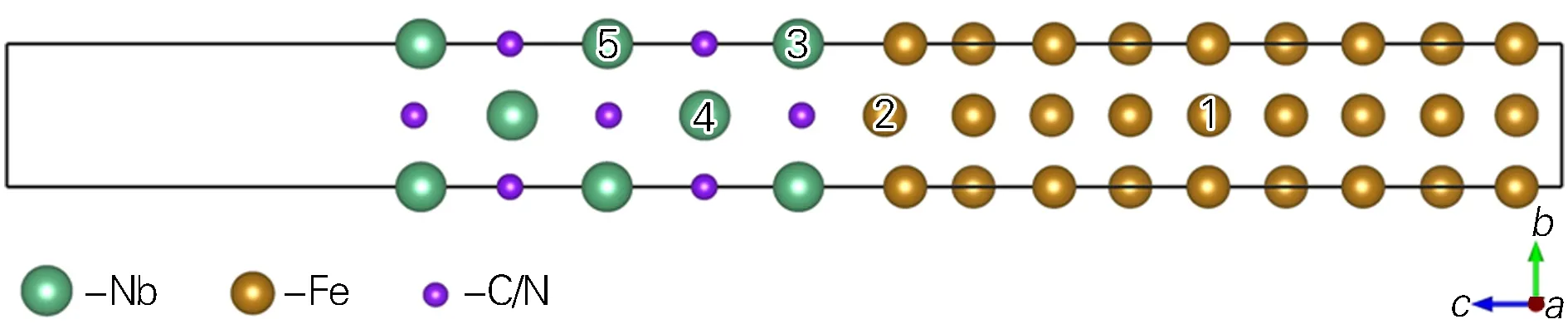
图1 fcc-Fe/NbX界面模型Fig.1 Model of fcc-Fe/NbX interface
偏析能(Eseg)用来表示替换原子M从基体到相界面偏析的倾向,由公式(1)确定:
(1)
其中:Einterface和Ebulk分别是替换原子M占据界面区域和基体区域界面模型的总能量。选处于原子层处的原子位点与界面平面和末端表面具有相等距离的位点作为基体位点。
格里菲斯断裂功常称为分离功,用来评估界面的结合能力(断裂强度)。通过添加替换原子M来预测内聚或内聚趋势,为将相界面分离为两个自由表面,即相界面断裂所需的能量,格里菲斯断裂功可通过公式(2)从相界面和两个自由表面的总能量之差得到:
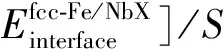
(2)
其中:S为界面处原子层的截面积,S=a×b,a和b为界面超晶胞的晶格常数;E(001)(NbX)和E(001)(fcc-Fe)分别为两个自由表面模型的总能量。
2 计算结果与讨论
2.1 fcc-Fe/NbC与fcc-Fe/NbN界面结构
通过计算fcc-Fe、NbC和NbN的晶胞和体积,验证了计算方法在这项工作中的准确性。表1为fcc-Fe,NbC和NbN的弛豫晶胞参数(a,b,c)和体积(V)。fcc-Fe,NbC和NbN的晶胞参数和体积与先前的试验和理论值一致[10-12]。平均偏差在计算允许范围内,表明计算中采用的参数可以确保足够的精度。
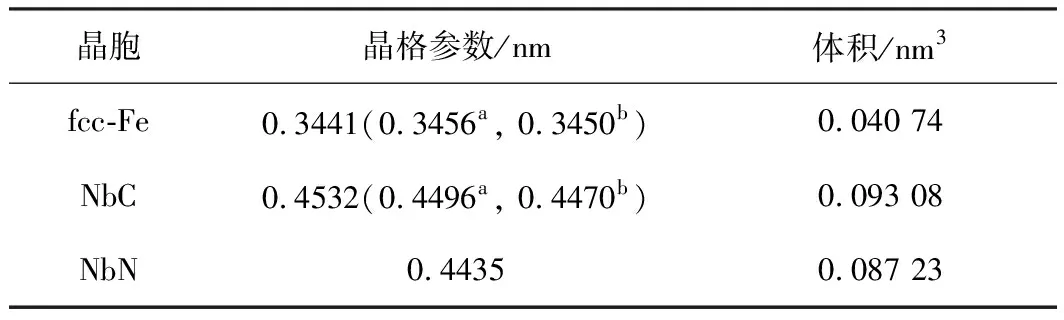
表1 fcc-Fe,NbC和NbN的计算晶格参数(a, b, c)和体积(V)
以此为基础,构建了两种类型的界面结构(fcc-Fe/NbC与fcc-Fe/NbN)。运用公式(2)比较了两种类型界面结构(fcc-Fe/NbC与fcc-Fe/NbN)的界面结合强度,表2为两种类型界面结构的界面各项参数及格里菲斯断裂功。可以发现(001)fcc-Fe/(001)NbN界面结合强度相较于(001)fcc-Fe/(001)NbC界面结合强度略有提升,这意味着NbN与fcc-Fe的结合更加紧密,后续将对其原子键合进一步分析。

表2 (001)fcc-Fe/(001)NbC和(001)fcc-Fe/(001)NbN界面的界面参数及格里菲斯断裂功
2.2 合金元素M界面处偏析倾向分析
根据公式(1)将替换原子M(Si、Ni、Mn、Cr、Mo)分别替换fcc-Fe/NbN界面结构体系中2/3/4/5位点的总能量记为Einterface;替换原子取代fcc-Fe(1位点)原子的界面总能量设置为Ebulk。图2为替换原子M在fcc-Fe/NbC、fcc-Fe/NbN界面不同位点(2/3/4/5)的偏析能。对图2(b)进行分析,可将替换原子界面偏析倾向分为3种类型:①替换原子Eseg值为正值,即这类原子存在于Fe基体中稳定,如Si。②替换原子处于界面处,其Eseg值为负值。这表明替换原子界面处轻微偏析,如Mn、Ni。③合金化界面的Eseg值均为负值。这表明替换原子界面处偏析,且存在进一步向氮化物中偏析的倾向,如Cr、Mo。
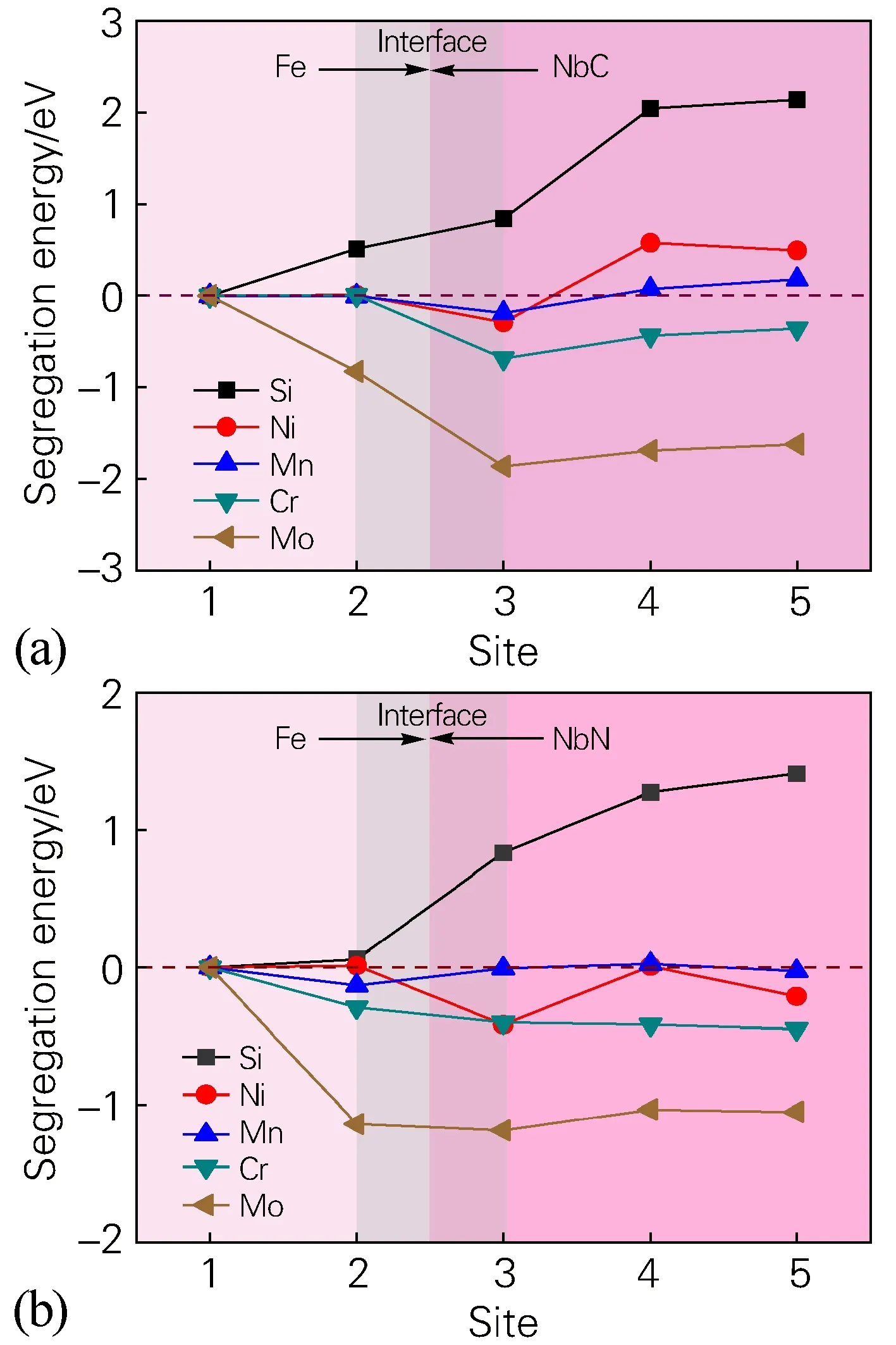
图2 合金元素位于界面不同位点(2/3/4/5)的偏析能(a)fcc-Fe/NbC界面;(b)fcc-Fe/NbN界面Fig.2 Segregation energy of alloying elements at different sites (2/3/4/5) on the interface(a) fcc-Fe/NbC interface structure system; (b) fcc-Fe/NbN interface structure system
将替换原子M在fcc-Fe/NbC界面不同位点的偏析能和在fcc-Fe/NbN界面不同位点的偏析能做比较,可以发现合金元素Cr、Mo在fcc-Fe/NbN界面处(2位点)的偏析倾向严重,但是在氮化物中偏析的倾向较在碳化物中偏析的倾向有所减缓。
为了进一步分析合金元素偏析能和合金原子的关系,绘制了合金元素偏析能和原子半径的关系图,如图3 所示。可以发现,除合金元素Mn外,合金元素偏析能(3位点)随合金原子半径的增大而变小,即偏析能的绝对值逐渐增大,偏析更加严重。
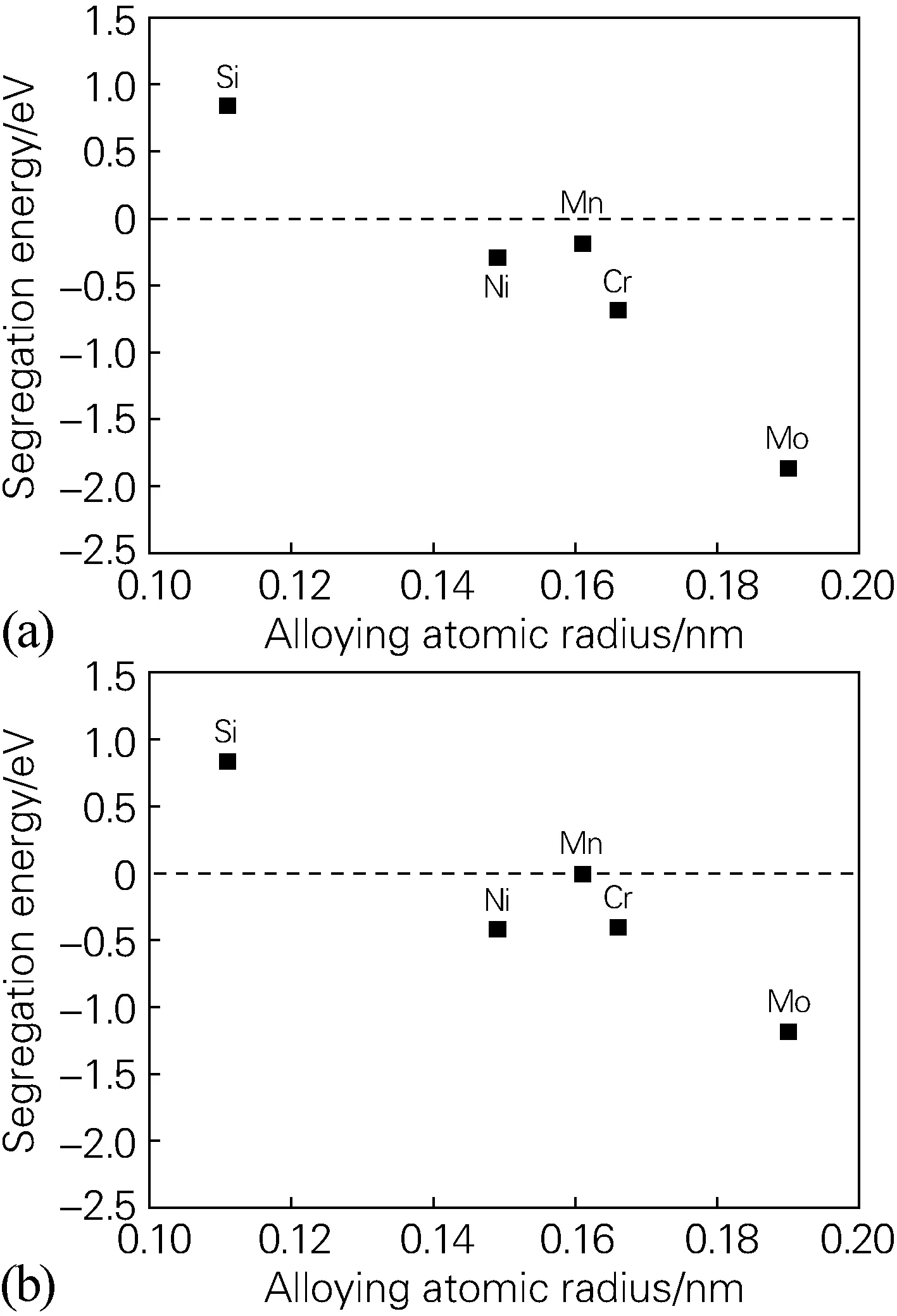
图3 合金元素位于界面3位点时的偏析能和原子半径关系(a)fcc-Fe/NbC界面;(b)fcc-Fe/NbN界面Fig.3 Relationship between segregation energy of alloying elements at 3 site of the interface and atomic radius(a) fcc-Fe/NbC interface; (b) fcc-Fe/NbN interface
图4(a,b)为替换原子M在fcc-Fe/NbC、fcc-Fe/NbN界面结构体系位于1、3位点时的格里菲斯断裂功,即Si、Ni、Mn、Cr、Mo等分别处于基体(1位点)、界面(3位点)后的界面分离功,其中用相应合金元素的偏析能着色这5种替换原子,因Si无偏析倾向,这里不予计算。对图4(b)进行分析,可以看出,替换原子处于Fe基体时,对界面分离功几乎没有影响(约为4.15 J/m2)。替换原子处于界面(3位点)时,除Ni外,界面分离功普遍高于替换原子位于Fe基体的分离功,说明替换原子处于界面(3位点)时提高了界面的结合能力。结合图2(b)偏析能,Mn在界面NbN侧无偏析倾向;而Cr、Mo原子更倾向于分布于界面的NbN侧,从而提高了界面的结合能力。对比fcc-Fe/NbC界面结构体系1、3位点的格里菲斯断裂功(见图4(a)),可以发现合金元素(除Ni外)偏析在fcc-Fe/NbC界面降低了界面的结合能力。
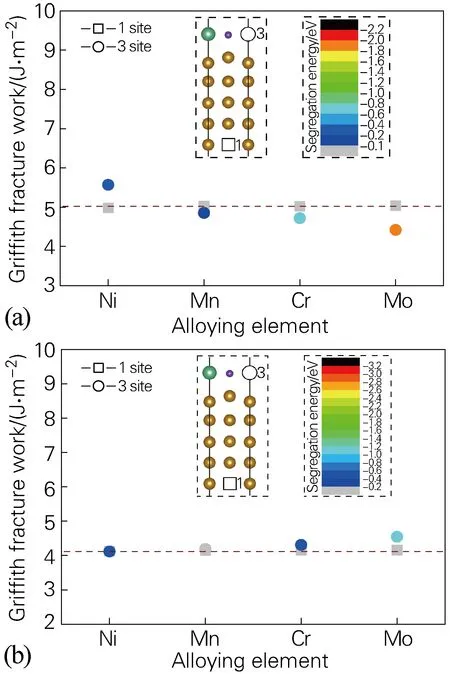
图4 合金元素位于界面1、3位点的格里菲斯断裂功(a)fcc-Fe/NbC界面;(b)fcc-Fe/NbN界面Fig.4 Griffith fracture work of alloying elements at 1 and 3 sites of the interface(a) fcc-Fe/NbC interface; (b) fcc-Fe/NbN interface
为了进一步分析合金元素位于界面时,界面体系的格里菲斯断裂功与合金原子的关系,绘制了合金元素分别位于界面结构体系1、3位点时,界面的格里菲斯断裂功和原子半径的关系图,如图5所示。可以发现,在fcc-Fe/NbC界面结构体系中,除合金元素Si外(因为Si不在界面处偏析),界面的格里菲斯断裂功(3位 点)随合金原子半径的增大而变小,即界面结合能力下降;在fcc-Fe/NbN界面结构体系中,界面的格里菲斯断裂功(3位点)随合金原子半径的增大而变大,即界面结合能力提高。
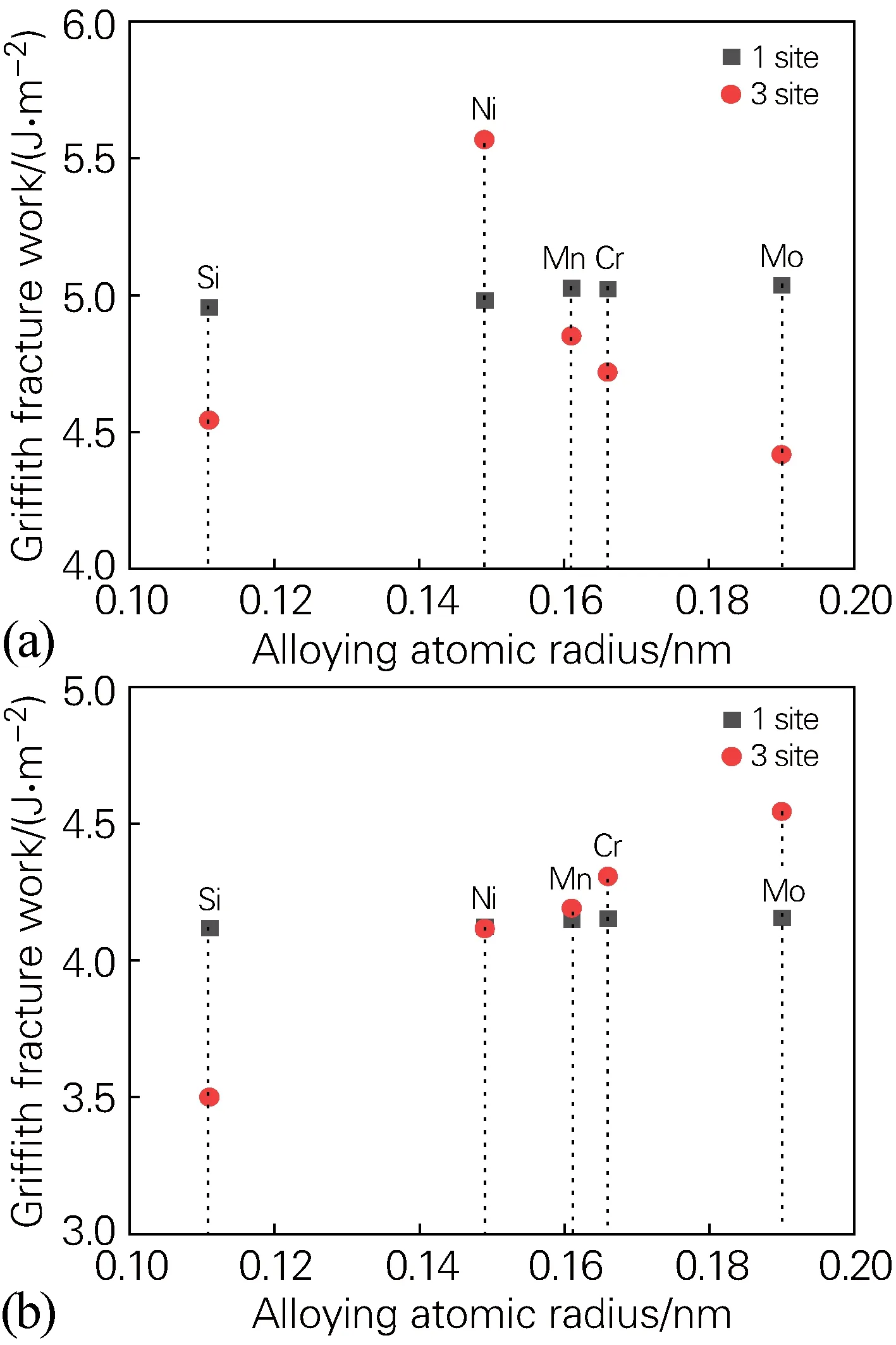
图5 合金元素位于界面1、3位点时格里菲斯断裂功和合金原子半径的关系(a) fcc-Fe/NbC界面;(b) fcc-Fe/NbN界面Fig.5 Relationship between Griffith fracture work and alloying atomic radius when the alloying elements at 1 and 3 sites of the interface(a) fcc-Fe/NbC interface; (b) fcc-Fe/NbN interface
2.3 合金化界面体系的电子特性
由于上述替换原子在界面时,对于界面结合能力影响不同,为了更为直观地理解替换原子与其最邻近原子之间的原子键合情况,对合金化界面附近相邻原子层的电荷密度进行分析。在每个合金化的界面中,Fe原子或Nb原子被特定的合金原子取代。因此,不同的替换原子M(M=Si、Ni、Mn、Cr和Mo)可能产生不同的结合强度。本部分合金化界面的Griffith断裂功通过将界面分成两个表面来计算。在微观层面上,这些界面的裂解断裂过程是破坏Fe-C(N)或M-C(N)键。因此,Fe-C(N)或M-C(N)键的键合强度对格里菲斯断裂有重要贡献。
以合金元素处于界面不同位置(1、3位点)为例进行计算,图6为合金元素(M=Si、Ni、Mn、Cr和Mo)分别处于fcc-Fe/NbC界面结构体系基体和界面处的电荷密度。可以发现,Fe基体内部的Fe原子周围的电荷呈圆形分布,不存在方向的极化分布。界面处Fe原子周围电荷分布存在一定方向性,而且与周围的C原子之间存在很大的电荷密度,结合Fe和C的电负性,可以认为Fe和C之间存在极化的共价性,且图6右边的竖直轴表示电荷密度值的尺度。同时,可以发现Si、Mn、Cr和Mo处于界面处时,界面原子间的电荷密度较其处于基体时的电荷密度小,这一趋势同前面叙述的合金化界面格里菲斯断裂功结果保持一致。
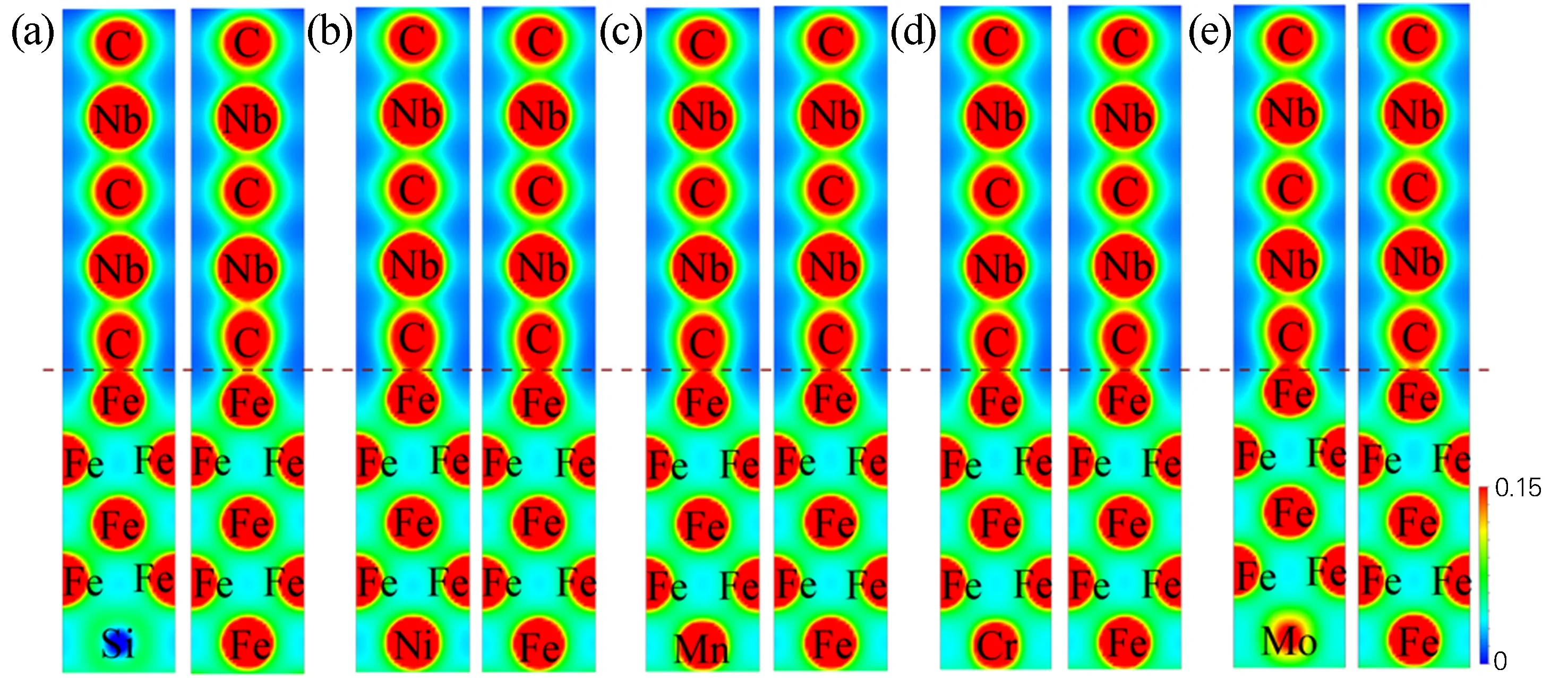
图6 合金元素位于fcc-Fe/NbC界面基体(1位点)和界面(3位点)处的电荷密度图Fig.6 Charge density diagrams of alloying elements at the matrix (1 site) and interface (3 site) of the fcc-Fe/NbC interface(a) Si; (b) Ni; (c) Mn; (d) Cr; (e) Mo
通过对纯净的界面体系和合金化界面(M= Si,Ni,Mn,Cr和Mo)体系态密度的分析,可以了解合金原子对界面体系的作用,进一步解释界面结合及结构稳定性。图7为纯净的fcc-Fe/NbC、fcc-Fe/NbN界面体系以及合金元素处于界面(3位点)时体系的态密度图。可以发现纯净的fcc-Fe/NbC、fcc-Fe/NbN界面体系费米能级处的总的态密度值分别为43.81、45.00 states/eV,表明fcc-Fe/NbC界面体系更加稳定。当在fcc-Fe/NbC界面体系处掺杂Cr、Mo时,费米能级处的态密度分别为42.42、41.25 states/eV,较之纯净界面体系有所降低,电化学活性下降,更加稳定。当在fcc-Fe/NbN界面体系处掺杂Cr、Mo时,费米能级处的态密度分别为44.78、48.26 states/eV,可以发现Mo的掺杂使得体系电化学活性提高,稳定性下降。
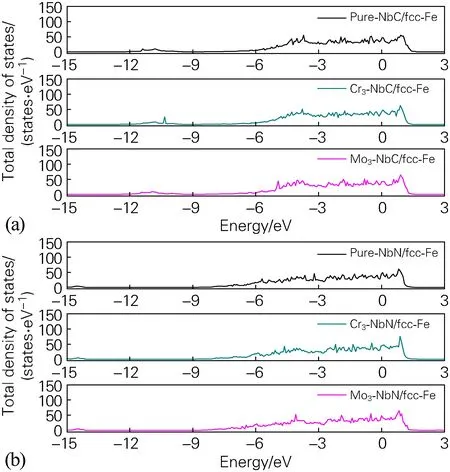
图7 纯净fcc-Fe/NbC、fcc-Fe/NbN界面体系以及合金化界面体系(3位点)的态密度Fig.7 Density of states of pure fcc-Fe/NbC, pure fcc-Fe/NbN interface system and alloyed interface system(3 site)
3 结论
基于密度泛函理论(DFT)研究了合金元素(M=Si、Ni、Mn、Cr、Mo)在fcc-Fe/NbX (X=C, N)界面的偏析行为、界面结合能力,并分析了合金元素对体系的影响。结论如下:
1) 在(001)fcc-Fe/(001)NbC界面体系中,Si更倾向于均匀分布于基体,Ni、Mn在界面NbC侧有轻微偏析的倾向,Cr、Mo处于界面和NbC均可稳定存在;在(001)fcc-Fe/(001)NbN界面结构体系中,Ni界面偏析趋势增大,Mn界面偏析扩展到界面Fe基体侧,且合金元素Cr、Mo在(001)fcc-Fe/(001)NbN界面的Fe基体侧偏析倾向更加严重。
2) 纯净的(001)fcc-Fe/(001)NbN界面结合强度高于纯净的(001)fcc-Fe/(001)NbC界面结合强度,这与C/N与Fe原子键和强弱有关;在(001)fcc-Fe/(001)NbC界面体系中,Cr、Mo在界面偏析降低了界面的结合能力,但提高了体系的稳定性;在(001)fcc-Fe/(001)NbN界面体系中,Cr、Mo在界面偏析提高了界面的结合能力,但Mo降低了体系的稳定性。
猜你喜欢
科技视界(2022年21期)2022-11-08
中国农业科学(2022年16期)2022-09-19
中国农业科学(2022年15期)2022-08-09
建材发展导向(2022年10期)2022-07-28
有色金属材料与工程(2021年5期)2021-10-25
速读·中旬(2021年10期)2021-10-14
电脑报(2020年40期)2020-11-06
科技视界(2019年27期)2019-11-05
有色金属材料与工程(2018年4期)2018-11-25
电脑知识与技术(2018年19期)2018-11-01
