
非酒精性脂肪性肝炎新药临床试验方案的系统评价
2022-04-14 04:21黄樱硕佟小非孙亚朦张健雄董瑞华贾继东
临床肝胆病杂志 2022年4期
黄樱硕, 魏 巍, 佟小非, 孙亚朦, 张健雄, 董瑞华, 贾继东, 尤 红
首都医科大学附属北京友谊医院 a.研究型病房, b.临床流行病学与循证医学研究室, c.肝病中心,d.肝硬化转化医学北京市重点实验室, e.国家消化系统疾病临床医学研究中心, 北京100050
非酒精性脂肪性肝病(NAFLD)是一种与胰岛素抵抗和遗传易感密切相关的代谢应激性肝损伤,疾病谱包括非酒精性肝脂肪变、非酒精性脂肪性肝炎(NASH)、肝硬化和肝细胞癌[1]。NAFLD和NASH已成为全球慢性肝病的最主要病因[2]。除了增加心血管疾病风险,NAFLD/NASH也是肝硬化、肝细胞癌和肝移植的主要病因之一[3]。已经有治疗NASH的多种新药临床试验处于早期临床研究阶段,但到目前为止,很多被寄予厚望的药物在2期和3期未达到预期的疗效终点,因此从NASH新药治疗的临床研发和临床试验的实施仍然面临巨大挑战。
目前NASH新药治疗的主要治疗靶点包括:法尼酯X受体(farnesoid X receptor,FXR)、成纤维细胞生长因子(fibroblast growth factor,FGF)、过氧化物酶体增殖物激活受体(peroxisome proliferators-activated receptors,PPAR)、抗炎/抗纤维化以及以改善糖脂代谢为目标的药物等[4]。
为了规范NASH新药研发和临床试验实施,更好地指导临床实践,2018年食品药品监督管理局(FDA) 发布了NASH伴肝纤维化治疗药物的研发指南草案[5],2019年美国肝病学会和欧洲肝病学会发布了对NAFLD临床试验终点的联合报告[6],2019年我国食品药品监督管理局发布了《非酒精性脂肪性肝炎治疗药物临床试验技术指导原则(试行)》[7],对NASH新药试验进行了详细的规范和指导。
本研究对国内外临床试验注册网站上关于NASH新药临床试验方案的筛选失败和试验失败原因进行系统分析,为今后NASH新药临床试验设计、招募和实施等过程提供参考和依据。
1 资料与方法
1.1 检索策略 (1)检索词。以Nonalcoholic Steatohepatitis为主题词检索美国临床试验数据库(www.clinicaltrial.gov),以“非酒精性脂肪性肝炎”和“NASH”为主题词检索中国临床试验注册中心(http://www.chictr.org.cn)以及国家药品监督管理局药品审评中心(http://www.cde.org.cn/),检索时间为建库以来至2021年8月6日。(2)检索条件。美国临床试验数据库检索状态选择包括:没有招募、招募中、通过邀请纳入、启动未招募、暂停、终止、完成;年龄组包括:成人、老年人;性别选择:全部;接受健康受试者;研究类型选择干预试验,研究分期包括:早期1期、1期、2期和3期。中国临床试验注册中心限定干预性试验;国家药品监督管理局药品审评中心不限定检索条件。
1.2 纳入标准 (1)研究类型:干预性试验;(2)研究对象:确诊为非肝硬化性非酒精性脂肪性肝炎的患者;(3)干预措施:NASH的新药,可合并肝纤维化,不伴代偿性/失代偿性肝硬化;(4)试验状态:尚未招募、招募中、通过邀请纳入、启动但未招募、暂停、终止、完成。
1.3 排除标准 (1)试验分期:Ⅳ期临床试验;(2)试验状态:已撤销、状态未明;(3)研究对象:仅诊断为NAFLD但未诊断为NASH的患者,其他代谢疾病如肥胖、糖尿病、心血管疾病患者,合并其他肝病或病毒感染(如乙型肝炎、丙型肝炎、丁型肝炎、HIV感染等)、合并代偿性/失代偿性肝硬化、合并肝功能异常(无论程度),儿童、青少年等非成人患者(<18岁);(4)干预措施:非NASH治疗新药,如不饱和脂肪酸、鱼油、维生素D、食物或其他营养素、干细胞、生活方式干预等;或不以改善NASH主要疗效指标作为评价标准,如以改善血脂或其他实验室指标作为主要评价指标等。
1.4 资料提取 由2位研究者独立查询和提取资料,并进行交叉核对,提取的信息包括:注册名称、官方名称、注册编号(NCT编号)、开始时间、试验状态(完成、终止、招募等)、试验分期、单一干预或联合干预、药物名称/药物代号、试验用药物作用机制(可通过Clinicaltrial.gov以外的其他渠道查询)、研究目的(安全性、耐受性、有效性、药代动力学、药效动力学等)、试验设计包括是否随机、是否盲法(包括开放标签、单盲、双盲、三盲、四盲)、有无安慰剂对照、干预类型(平行试验、序贯试验、交叉试验、单臂试验)、是否适应性设计、入组例数、主要入选标准(病理、影像学、临床诊断、ALT、BMI限制范围、有无体质量变化的限制、主要结局评价指标(安全性、耐受性、病理改变及间隔时间、影像学改变及间隔时间、ALT改变及间隔时间)类型、申办方所在国家、是否单中心/多中心研究等。(3)由2位研究者独立检索及筛选文献,意见不一致时进行讨论或参考第3位研究者的意见。
2 结果
2.1 注册情况及基本情况分布 共检索到符合上述条件的NASH相关注册试验共400项(美国临床试验数据库354项,中国临床试验注册中心21项,国家药品监督管理局药品审评中心25项),除外仅以NAFLD作为研究对象、合并肝纤维化、非NASH的其他代谢性疾病、合并其他病毒感染(如HCV、HIV感染)、不以NASH作为治疗目标、非药物干预(营养成分/食物/其他生活方式)、合并肝肾损伤、药物相互作用、物料平衡、儿童等情况,以及国内外多中心针对同一药物的分别注册的试验,共筛选出196项(图1)。
2.2 试验地域分布及完成情况 根据本研究的入排标准,最后纳入的为196项。申办方为中国(含台湾地区)企业共有38项,其他国家共158项,以美国最多,为105项。按照中心数量分组,包括多中心研究104项,单中心研究92项(图2)。
试验进行状态分布情况:启动未招募25项,完成 97项,通过邀请入选 1项,招募过程中 53项,暂停3项,终止10项(终止原因包括安全事件、缺少预计疗效、未达主要疗效终点、根据卫生部门意见、申办方决定、实验室异常值等),状态不明7项。详见图3。
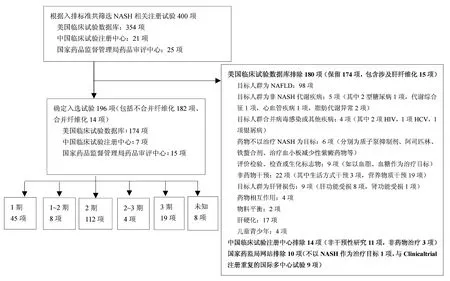
图1 筛选流程图
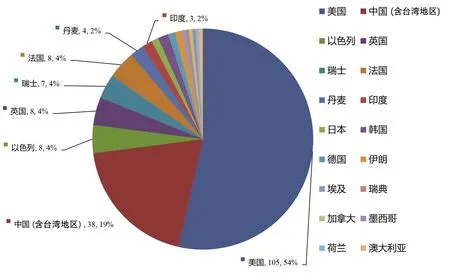
图2 申办方公司所在国家的注册试验数量Figure 2 The number of clinical trials in different countries where sponsor companies located
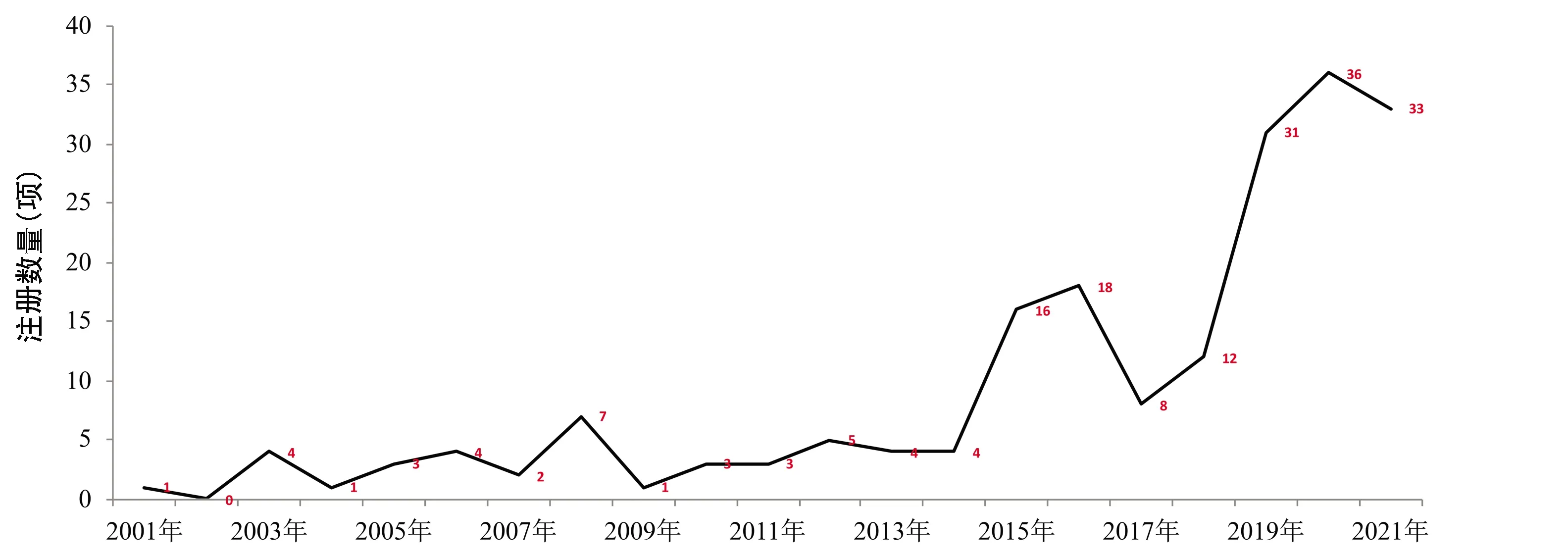
图4 注册试验数量随年份分布情况Figure 4 Numbers of registered trials distributed by year
2.3 试验时间分布情况(按照注册时间或开始时间计算) 2000年以前没有NASH注册试验,2001年—2005年共有9项,2006年—2010年共17项,2011年—2015年共32项,2016年—2020年共105项,2021年到筛选日为止33项,注册数量呈递增趋势(图4)。
2.4 NASH治疗药物机制类型和给药类型 NASH治疗药物主要包括如下种类:FXR类16项(激动剂14项,受体抑制剂1项,受体配体1项),FGF类14项(FGF受体抗体3项,FGF-21类似物8项,FGF-19类似物2项,FGF激动剂1项),PPAR激动剂11项,胰高血糖素样肽(glucagon-like peptide,GLP)-1类13项(激动剂9项,类似物4项),胰岛素增敏剂7项,激素类6项(睾酮类3项,糖皮质激素类2项,雌激素类1项),保肝药5项(水飞蓟素类3项,胆汁酸调节剂2项),己酮可可碱类似物4项,趋化因子受体(chemokine receptor,CCR)拮抗剂4项,乙酰辅酶A(acetyl CoA carboxylase,ACC)抑制剂3项,脂肪酸合成酶抑制剂3项,胰岛素3项,瘦素类似物3项,抗生素3项,大麻素受体抑制剂3项,降脂药3项(依折麦布1项,考来维仑1项,阿托伐他汀1项),钠-葡萄糖协同转运蛋白(sodium-glucose-linked transporter,SGLT)抑制剂3项,顶端钠离子依赖性胆酸转运体抑制剂2项,酮己糖激酶抑制剂2项,氨基脲敏感性胺氧化酶抑制剂2项,其他46项[其中包括利尿剂1项,Toll样受体4拮抗剂1项,甲状腺激素受体β1(激动剂1项,磷酸二酯酶4抑制剂1项,RNA干扰(RNA interference,RNAi)类药物1项],各种中药10项,未知30项(见图5,仅列举数量较多的靶点类型,数量较少的靶点类型合并为其他新药机制)。比例最高的前4名依次为FXR类16项(8.16%),FGF类14项(7.14%),PPAR激动剂11项(5.61%),GLP-1类13项(6.63%)。其中单药给药为180项(91.8%),联合给药为16项(8.2%)。
2.5 NASH新药临床试验的设计特点 按照是否随机分为随机试验和非随机试验,其中非随机试验23项,随机试验173项;按照盲法分类包括开放标签35项、单盲6项、双盲53项、三盲26项、四盲75项、未知1项,其中采用盲法为112项(57.1%)。163项试验采用了安慰剂对照,2项涉及生活方式干预作为对照,3项阳性对照。
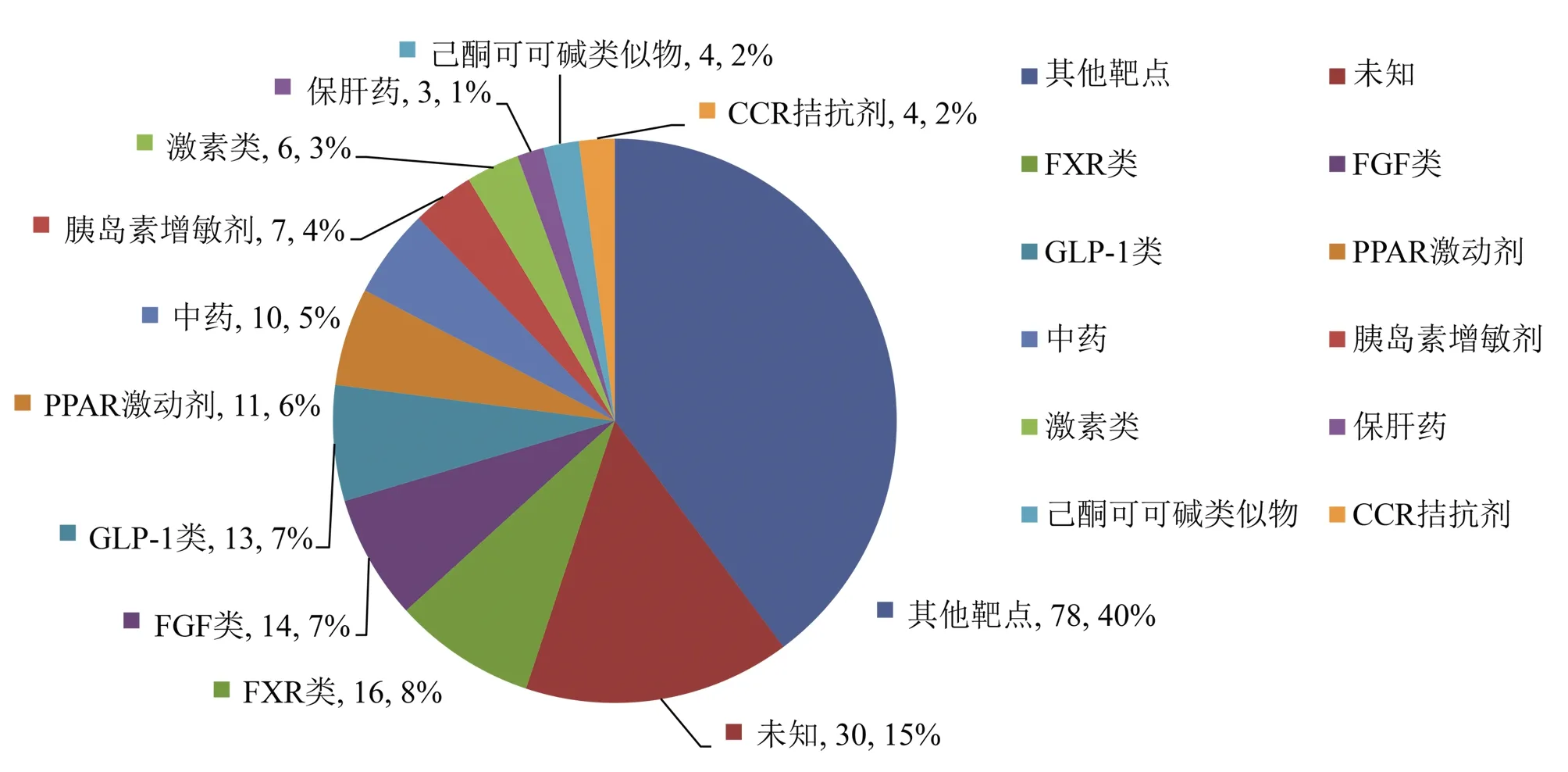
图5 NASH新药试验的靶点类型Figure 5 Types of targets for NASH drug trials
根据干预模型分类,平行试验159项,序贯试验12项,单臂试验18项,交叉试验4项,未知3项。
根据是否新型试验设计分类,常规设计有193项,适应性设计有3项。
根据试验评价目的分类,其中目的涉及安全性有145项,涉及耐受性77项,涉及效果138项,涉及药代动力学/药效动力学有62项(上述研究目的部分有重叠)。
2.6 NASH新药试验的主要入选标准
2.6.1 病理作为主要入选标准 以NASH作为受试对象的试验中,病理作为主要入选标准共有125项,占全部试验的63.8%,其中病理诊断NASH的有效时间范围,接受病理有效时间分为3个月、6个月(含18~24周)、12个月(含1年、360 d、48周)、18个月、24个月、36个月,分别为3项、39项、21项、2项、7项、2项,另外有51项尽管有病理作为入选标准,但对病理结果的有效时间没有规定;病理有效时间为6~12个月的临床试验数量最多(图6)。对于病理评分,主要以NAS评分和NASH-CRN病理评分的分级为标准[8],其中≥4分为35项,≥3分为6项,≥5分为2项,并且规定小叶炎症和气球样变等不同维度均≥1分;按照NASH-CRN评分系统的分级,F2~F3为15项,F1~F3为12项,F1~F4为2项。
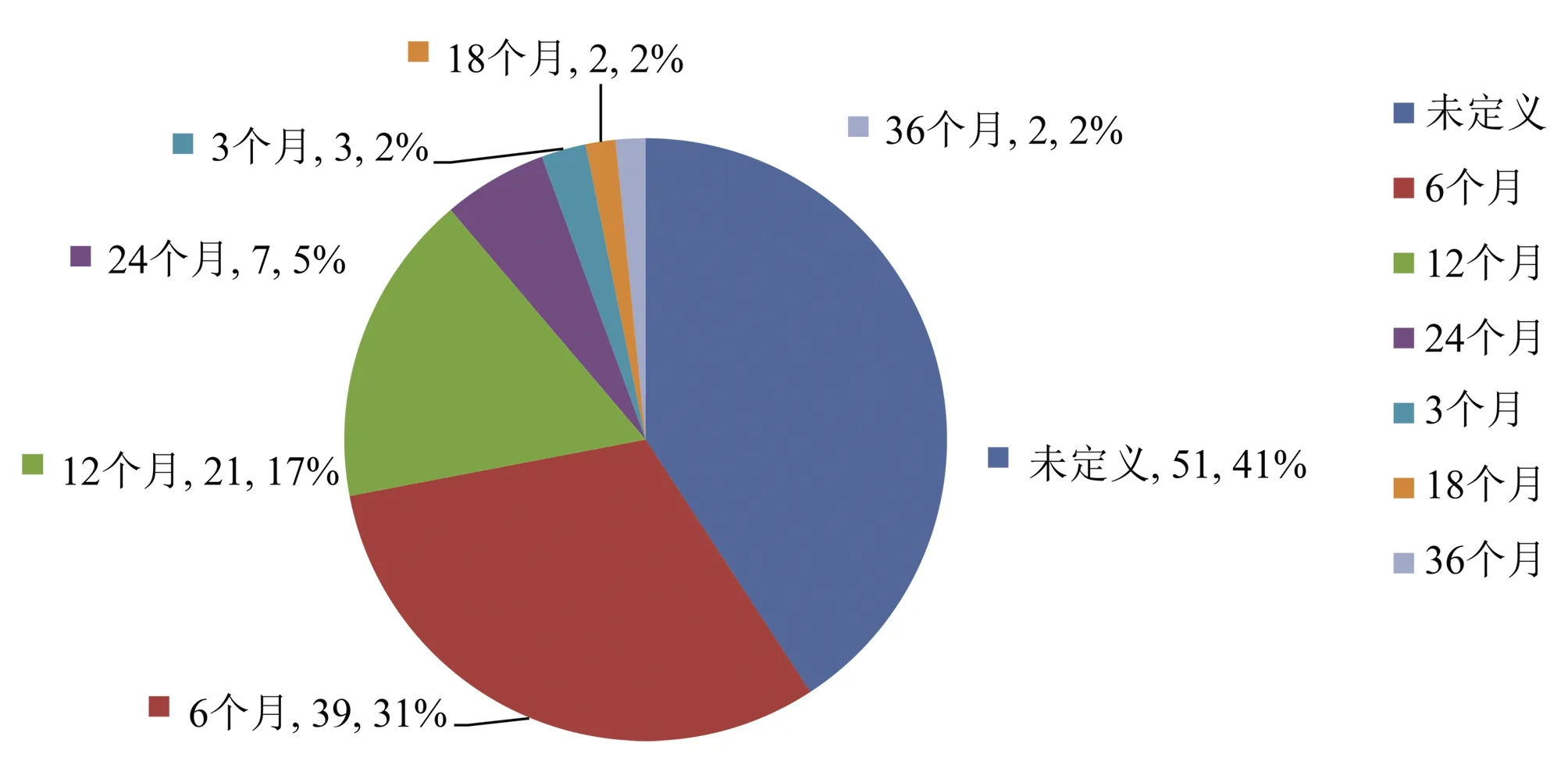
图6 NASH新药试验病理评价诊断标准的有效时间Figure 6 Effective time of pathological assessment for NASH drug trials
2.6.2 影像学作为主要入选标准 以NASH作为受试对象的试验中,影像作为入选标准共有66项,占全部试验的33.7%,其中以MRI质子密度脂肪分数(proton density fat fraction,PDFF)作为入选标准有46项,定义为≥8%(含>8%和≥7%)有21项,≥10%(含>10%)有15项,≥5%(含>5%及≥5.5%)有5项,>15%有1项;对磁共振弹性成像(magnetic resonance elastography,MRE)有规定的有8项,范围为2.5~3.63 kPa;以Fibroscan作为入选标准的有21项,受控衰减参数(CAP)>230~300 dB/m,肝脏瞬时弹性值/肝脏硬度值(LSM)为7~13 kPa。
2.6.3 临床诊断作为主要入选标准 在非Ⅰ期研究中(包含Ⅰ~Ⅱ期),以临床诊断作为入选标准的有42项,占全部试验的21.4%。其中对ALT范围进行规定的有22项。
2.6.4 对BMI和体质量变化的规定 对BMI有范围规定的有98项,其中34项仅对BMI低限进行规定,10项仅对BMI高限进行规定,54项同时对BMI的低限和高限进行规定;BMI低限≤24 kg/m2有40项,25~28 kg/m2有40项,>28 kg/m2有8项;BMI高限≤28 kg/m2有13项,29~35 kg/m2有18项,>35 kg/m2有33项。对体质量变化有限制规定的59项,其中增加或减少以≤5%为界有28项,10%有21项(含7%1项),5 kg为界的2项,未规定7项;体质量稳定时间范围≤3个月有19项,6个月有22项,12个月有6项,未规定有12项。
2.7 NASH新药试验的主要评价指标 主要评价指标与试验分期有关,其中Ⅰ期主要以安全性、耐受性及药代动力学参数为主要评价指标,Ⅱ~Ⅲ期试验除评价安全性,还评价有效性,NASH新药试验的有效性主要以病理、影像学的变化为主要指标。在入选的全部试验中,以安全性为主要评价指标有120项,以耐受性为主要评价指标有78项,药代动力学指标为主要评价指标有19项。以病理学为主要评价指标有62项,病理评价的有效时间≤6个月(含26周)、28~52周、72~120周对应的试验数量分别为14项、34项和13项,还有1项试验未规定病理评价的有效时间。以影像学改变为主要评价指标的试验有40项,观察时间为≤24周和28~52周分别有31项和6项,还有3项未规定观察时间。以ALT改变作为主要观察指标的试验有28项,观察时间为≤24周和28~52周分别有20项和4项,其他4项未规定观察时间。
3 讨论
NASH新药数量呈递增趋势,尤其是近5年,较前成倍增长,表明申办方和研究者对NASH的认识不断深入,对NASH治疗的需求也在增加,除了已完成的100项试验,还有进行中的近80项试验。由于发展至肝硬化阶段的NASH与非肝硬化性NASH在治疗上有较大差异,因此本文在进行分析时排除了肝硬化,仅纳入非肝硬化性NASH。
根据申办方的地区分布,仍以欧美国家为主,其中美国数量最多,在非欧美地区,中国的申办方注册试验的数量最多,并且有多项1.1类新药,还有部分为中药。国内注册试验数量近年来也有逐渐增加的趋势。随着中国加入国际人用药品注册技术协调会,NASH新药发展速度也进入了快车道。
NASH/NAFLD新药治疗以改善临床结局、代谢结局、炎症和纤维化等作为目标,新药设计的靶点主要包括改善代谢和胰岛素抵抗、抗炎、抗纤维化等作用于NASH的不同机制[9]。数量最多的药物涉及FXR类、FGF类以及PPAR激动剂、GLP类、CCR和ACC抑制剂等[9-10],除了GLP类为新型降糖药,还有传统的降糖药、降脂药、激素类药物也在开发关于NASH的新适应证[4]。中国自主研发的NASH新药仍以化学药品为主,还有一部分中药试验。除了国外申办方发起的国际多中心试验在中国进行注册外,中国自主研发的NASH新药的作用机制几乎未公开,与国际NASH新药的作用机制无法比较。
NASH新药试验以单药治疗为主,因为研究对象大部分为病理分级在F3以内的人群,部分试验涉及纤维化,少数试验为联合治疗。联合用药也是近年来NASH新药试验发展的一个趋势,如GLP-1类似物与ACC抑制剂、FXR激动剂的双药或三药联合,白三烯A4水解酶抑制剂与FXR激动剂的联合,FXR激动剂与阿托伐他汀联合,FXR激动剂与SGLT抑制剂联合,FXR激动剂与趋化因子受体CCR2/5双重拮抗剂联合等。这些联合方案以不同NASH治疗的不同机制出发,通过抗炎、抗纤维化等效果联合实现治疗NASH的目的,这也是NASH新药治疗试验设计的一种新模式和新思路。
NASH新药试验多为随机、盲法、安慰剂对照的试验设计,其中双盲、三盲、四盲的比例最高,开放标签和单盲的试验较少,从研究设计类型看,主要涉及平行试验、序贯试验、单臂试验、交叉试验这四种类型,其中平行试验占大多数,是最常用的临床试验设计类型。由于NASH尚无公认有效的治疗药物,目前一般以安慰剂作为阴性对照,少数以生活方式干预作为对照。大多数NASH新药试验处于早期探索阶段,大多因为没有达到预期终点而未能成功上市。尽管新型试验设计有望减少研究成本,但由于NASH新药试验的结果具有较大的不确定性,总体而言现有的试验仍以传统设计为主。
NASH的诊断仍以病理作为金标准,但是随着对NASH认识的增加,以及临床试验数据的支持,MRI-PDFF、MRE、Fibroscan等无创影像学手段在相对早期阶段的NASH试验的地位逐渐提升[11],而且作为金标准的病理诊断,可能存在较大的组间和组内诊断差异[12],但对于确证性试验,病理仍作为最重要的入选和疗效标准。根据美国FDA、美国肝病学会和欧洲肝病学会、我国食品药品监督管理局发布的相关指导文件,可将病理、影像学、临床表型诊断作为主要的入选标准。由于NASH可能受到生活方式、体质量变化等的影响,病理状态、评分和CRN-NASH分级也可能在一定时间内发生变化,因此比较规范的NASH新药试验对病理有效时间和体质量变化范围及时间均做了明确的规定,占比最高的病理有效时间为6~12个月,但仍有较多试验对病理的有效时间没有做出明确规定。
目前主要以NAS评分作为NASH的主要病理评价标准[8],NAS评分包含脂肪变性、小叶炎症和气球样变三个维度,除了规定NAS评分外,还对每个维度的得分进行限制,多数为小叶炎症和气球样变的不同维度得分均≥1分,这样NASH的病变较均衡。对既往病理有效时间的规定范围与试验分期有关。大部分试验接受12个月以内,以3~6个月为主。对于12个月以上的病理有效时间,或者为早期试验,或者在入选标准中还结合了影像学标准、代谢状态等更多限定条件。
对以降低脂变为治疗靶点的NASH新药中,MRI-PDFF是一种比较好的无创影像学评价方式[13],可以在一定程度上代替病理金标准,有研究[14]表明影像学与病理的一致性较好。目前MRI-PDFF诊断NASH的标准比较统一,多数认可8%~10%的范围,但MRI-PDFF也存在组间判断差异较大的问题[15]。MRE受到仪器和人员的限制,开展和适用性受限,无法普遍实施。Fibroscan是近年来新发展的无创检测手段[16],但是受到检查者的操作因素影响较大,并且注册试验中, CAP和LSM限定值的变化范围也较大,可能需要更多对比数据支持。
虽然NASH与肥胖的关系较为密切,但是仍有部分瘦型NASH人群,一般来说,Ⅰ期试验以健康受试者为主,对BMI多要求在正常范围,而Ⅱ、Ⅲ期试验纳入多为NASH患者,因此BMI限定超重和肥胖患者的试验数量较多,但是在试验设计时不能忽略瘦型NASH这一特殊人群,可以适当扩大BMI的入选范围,以病理及影像学的诊断为主。体质量变化可能对NASH的新药疗效、观察指标等都有影响,也可能导致安慰剂效应[17-18]。因此较多试验的入选标准对体质量变化进行了规定,要求在筛选至基线期保持稳定。
Ⅰ期试验的受试对象为健康人,主要以安全性、耐受性和药代动力学指标为主要评价重点;对于Ⅱ期NASH新药试验的疗效评价,以病理和影像学作为主要疗效评价标准[11,19-20]。疗效评价的病理观察时间以半年至1年为主,而影像学的变化以半年内为主。Ⅲ期试验,大部分将病理组织学的改善作为替代终点[11],因为NASH发展至肝硬化甚至出现死亡事件的时间较长,常规的Ⅲ期试验可能较难观察到临床结局事件。部分观察时间较长(如5年)可能纳入了肝硬化、死亡等临床终点事件。对于观察指标的时间范围主要与试验分期、研究目的等有关。
由于NASH新药试验的观察时间较长,因此导致新药研发周期较长;尽管很多新药试验将血清生化标志物如细胞角蛋白18[9,20]或一些多项复合指标的评分系统如增强的肝纤维化评分等[21]也作为评价指标,但由于新药作用机制的多种多样,目前并无公认统一的生化标志物可供作为疗效评价指标。探索能够早期诊断和评价NASH疗效的指标也是新药试验的研究方向之一。
综上,国际NASH创新药临床试验注册量增长速度较快,近年来显著增多,多处于早期阶段,药物作用机制多种多样,入选标准和评价指标差异较大,病理和影像学仍为主要的入选标准及疗效评价指标,联合治疗和纳入代谢状态作为复合指标可能是是未来试验设计的趋势,但病理和影像指标的具体值及观察时间缺少统一的标准。由于观察时间的限制,临床事件硬终点的支持证据相对较少,并且新型试验设计较少。国内注册的NASH新药临床试验尽管数量较少,但总体数量呈增长趋势;相对化学类新药,中药注册试验数量较少。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:尤红、贾继东负责选题及文章结构设计;黄樱硕、魏巍负责检索数据库、筛选文献及提取资料;黄樱硕、佟小非负责撰写文章并修改;董瑞华、孙亚朦、张健雄负责对结果进行分析和讨论;黄樱硕参与完成初稿的撰写。
猜你喜欢
华人时刊(2022年1期)2022-04-26
中国典型病例大全(2022年7期)2022-04-22
安徽农业科学(2022年6期)2022-04-11
健康体检与管理(2021年10期)2021-01-03
中国当代医药(2020年6期)2020-04-01
家庭医药(2020年2期)2020-03-17
文萃报·周二版(2019年8期)2019-09-10
祝您健康(1991年5期)1991-12-29
