
高通量单细胞转录组数据分析方法的研究进展
2022-04-14 12:48张泽坤顾连峰
福建农林大学学报(自然科学版) 2022年2期
李 涛, 张泽坤, 顾连峰
(1.福建农林大学林学院;2.海峡联合研究院基础林学与蛋白质组学中心,福建 福州 350002)
自2009年首个单细胞转录组测序(single-cell RNA sequencing, scRNA-seq)技术发布以来,scRNA-seq被越来越广泛地应用于基础科学研究中,特别是在发现新的异质细胞类型和追踪细胞动态发育轨迹中发挥了重要作用.目前,scRNA-seq的通量有了质的提升,由原来的几个细胞发展到成千上万个细胞.通量增加的同时也提高了数据的复杂性,使后续数据分析面临巨大挑战.大多数分析软件缺乏图形界面且需要研究者设置参数,不同的参数选择会导致截然不同的分析结果.而怎样衡量这些由参数变化引起的不同结果,及其是否具有显著的生物学意义在行业中仍然缺少标准.此外,scRNA-seq所产出的数据相比于普通转录组测序(bulk RNA sequencing, Bulk RNA-seq)存在一些独特的性质,例如,由于单个细胞的RNA含量在皮克(pg)水平,scRNA-seq测序的数据集具有更高的噪音水平和更多的零值(缺失值)[1].缺失值的产生不仅来源于技术噪音,还有可能具有生物学意义,即该基因在细胞中并未表达.因此,对高通量scRNA-seq数据的分析流程(图1)和常用软件的介绍可为数据研究提供合适的方法.
1 单细胞转录组数据的质量控制和标准化
1.1 数据质量控制
获得测序数据后的第一步是进行预处理和质量控制,早期常用的预处理和质量控制工具包括Trimmomatic[2]、Cutadapt[3]、Kraken[4]等.这些软件开发之初并非是针对scRNA-seq所设计,无法适应scRNA-seq数据集技术噪音(technical artifacts)和内在生物学变异相互结合的复杂特征.有研究表明,可通过管家基因[5,6],基因的整体表达模式[7,8],以及检测到的基因数量和(或)读段比对的比例去除技术噪音[9],但是这些方法均存在一定程度的局限,可能会过滤掉一些内在的生物变异.随着scRNA-seq的广泛应用,开发了专门针对scRNA-seq数据集的质量控制软件,SinQC工具可以通过综合基因表达模型和文库质量信息去除技术噪音[10].除了专门的质量控制工具以外,Scater等综合性工具内也集成了质量控制和预处理组件[11].完成质量控制后的下一步是进行序列比对,常用的比对程序是已在Bulk RNA-seq中广泛应用的STAR[12]、BWA[13]等软件.有些综合软件已经内置了比对组件,例如10X Genomics公司的Cell Ranger内置了STAR作为比对组件,可以一次性完成基于10X Genomics chromium产出的单细胞数据的上游分析.对于在试验中应用了单分子标签(unique molecular identifier, UMI)进行表达量校准的协议,应该在比对之前移除这些序列.
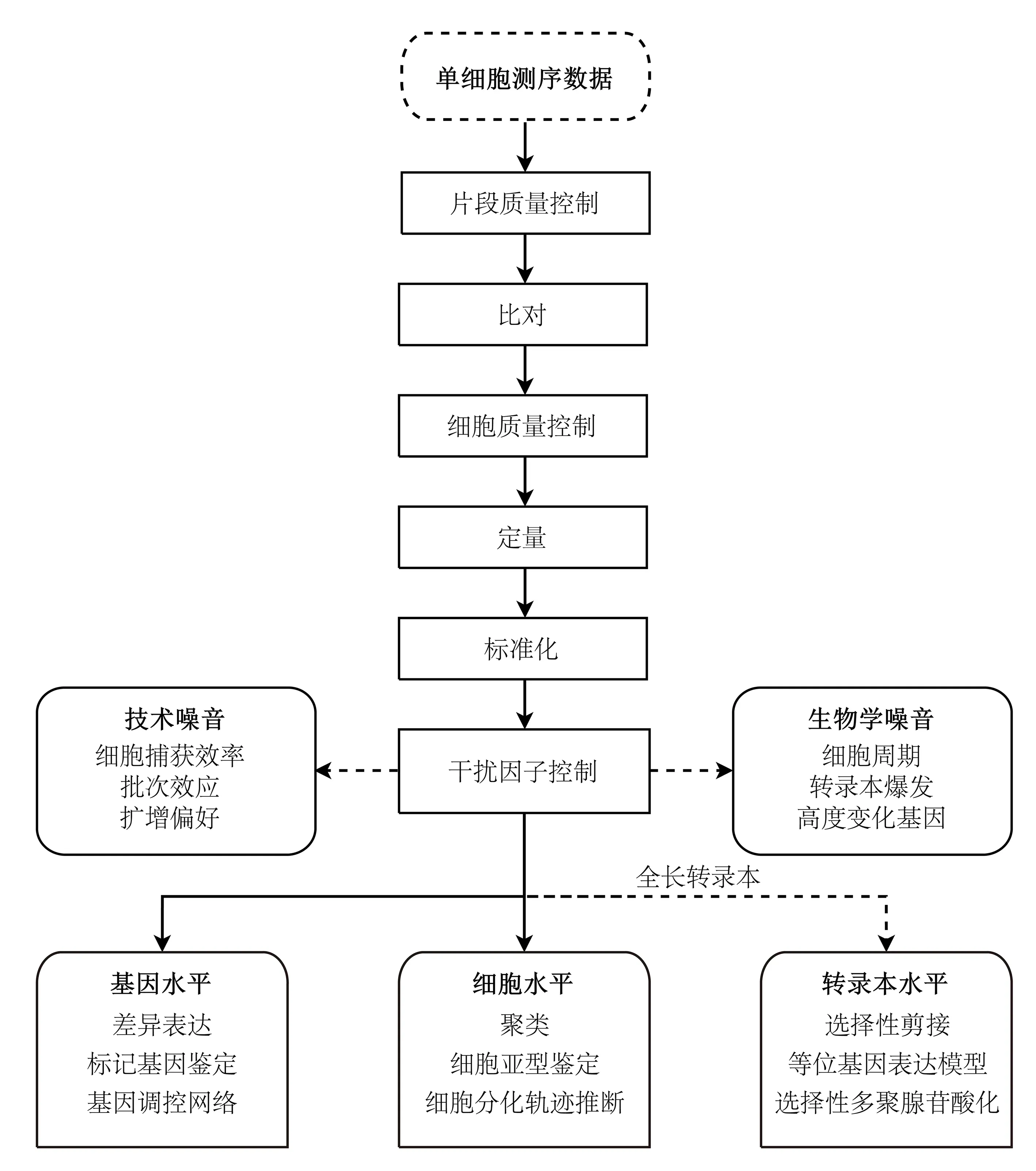
图1 单细胞转录组生物信息学分析流程Fig.1 Schematic overview of bioinformatic analysis workflow for scRNA-seq
过滤掉低质量的细胞对于有些scRNA-seq协议也是十分重要的,现有的许多scRNA-seq协议在捕获细胞时可能会对细胞造成损伤甚至死亡,在一些“液滴”中还可能会包括多个细胞或者未捕获到细胞.这些低质量的细胞数据应当在进行下游分析之前被剔除,避免造成最终结果的失真.Ilicic et al[14]提出了基于一组通用特征合并机器学习算法识别并去除上述低质量细胞的工具.有时,内参(spike in)也被用作质量控制的一种常用手段,当内参序列比对到参考基因组的比例较高时,提示该细胞的质量较低,其内源RNA被降解或者细胞裂解不够充分.在低通量的协议中,显微成像是重要的质量控制手段,通过对细胞的完整性进行检测以剔除一些破碎或死亡的细胞,但是当被测序的细胞数量达到数以万计时,对每个细胞进行显微成像变得不再可能.
1.2 标准化
标准化(normalization)是scRNA-seq数据分析中关键的一步,将直接影响下游分析结果的可靠性.目前常用的标准化方法大多来自于Bulk RNA-seq,分为两类,一类基于缩放,例如reads per million (RPM)、fragments per kilobase of transcript per million mapped reads (FPKM)、trimmed mean of M values (TMM)[15]和 DESeq[16]等.这些方法并不适合日趋大型而复杂的单细胞数据集,处理批次效应和零膨胀等问题时可能会引入误差[17].另一类是像remove unwanted variation (RUV)、surrogate variable analysis (SVA)[18]这种需要调整关键参数的方法.大多数在Bulk RNA-seq中应用的标准化方法都基于一个内在的假设,即每个样本中处理的RNA总量相似或者其变化是技术引起的.但是,这个假设在某些情况下是不可靠的,可能会使下游分析失真[19,20].随着单细胞测序的发展,逐步开发了一系列适用于scRNA-seq数据集的标准化方法,下面简要介绍这些方法.
(1)不基于内参的标准化:标准化的选择主要基于所采用的scRNA-seq协议的特征,在某些协议中内参的使用是困难的,例如,一系列基于“液滴”的协议[21-23],无法有效地利用内参进行标准化.Lun et al[24,25]提出了一种反卷积方法,通过对基因表达值进行跨细胞求和,并对求和后的值进行标准化,然后利用反卷积将基于跨细胞的因子(pool-based size factors)转化为细胞层次因子(cell-based factors),从而有效地减少单细胞数据集零膨胀导致的失真.该方法已经被封装到R包scran中,可以通过调用computeSumFactors函数实现反卷积标准化.Qiu et al[26]提出了Census算法,通过将传统的相对表达量度(例如FPKM)转换为单细胞水平的转录本相对计数,在无需内参和UMI的情况下,改善下游分析的准确性.Census算法还可以结合回归分析用于对细胞命运分支点的调控,单细胞差异剪接和等位基因平衡的研究,可通过调用R包Monocle 2使用前述功能.但是值得注意的是,Census算法不能有效地控制扩增偏倚,所以在需要准确计算RNA丰度时,应选择含有内参或(和)UMI的协议.Hafemeister et al[27]开发了R包sctransform,提出了针对UMI协议数据集的标准化方法.sctransform使用正则化负二项分布的Pearson 残差,在保留生物异质性的同时消除了技术差异的影响.不同于常用的标准化策略,sctransform的工作流程中并未使用缩放因子,而是专注于为每个基因构建单独的广义线性模型,使用UMI计数作为因变量,测序深度作为解释变量,计算该模型的Pearson 残差,消除基因平均表达和细胞变异之间的固有依赖,从而有效地去除技术差异的影响.sctransform目前已被直接集成到单细胞分析R包Seurat中,可以直接在Seurat中调用相关函数.
(2)基于内参的标准化:定量每个细胞中的总RNA 含量,对于scRNA-seq的下游分析十分重要,而这点无法利用全局缩放准确做到.Jiang et al[28]尝试了在Bulk RNA-seq中使用人工合成的内参进行定量,在一些scRNA-seq试验中内参也被尝试使用[29-32].内参基因被添加到每个细胞中并且在所有细胞中处于相同水平,故可以通过对内参基因与细胞自身基因表达水平的比较定量技术噪音和总RNA量.目前被广泛使用的内参是来自于external RNA controls consortium (ERCC)的92个外源分子集合[28].一些基于内参对scRNA-seq数据进行标准化的综合工具被开发.例如,GRM[30]、BASICS[32]、SAMstrt[31]等.GRM通过对测序读段的表达量(RPKM, FPKM或TPM)和掺入的ERCC进行伽马回归模型拟合,以消除技术噪音并计算基因表达量.GRM是对样本内基因表达水平进行标准化的工具,不能用于样本间的标准化[17].BASICS基于贝叶斯分层模型整合细胞自身基因和ERCC信息以量化无法解释的技术噪音和细胞间的异质性.
(3)特殊的标准化:SCnorm是样本间的标准化方法,使用分位数回归对测序深度具有相似依赖性的基因进行分组,并为每组估计不同的缩放因子,然后使用估计的比例因子对测序深度进行组内调整,以达到标准化的效果.SCnorm是为了解决全局缩放时可能导致的弱表达和中等表达基因被过度校正而开发,既可以对无内参的scRNA-seq数据进行标准化,也可以对有内参的scRNA-seq数据进行标准化.当内参均匀分布在整个内源基因中时,可以有效改善标准化的性能[33].scLVM(single-cell latent variable model)可用于对细胞周期和其它混杂因素引起的变异进行标准化.同时,scLVM也允许使用者对预定基因集的潜在因素进行建模和解释,从而研究各种scRNA-seq试验的变异来源[29].
大部分使用内参进行标准化的方法都基于一个重要的假设,即生物协变量不影响内参序列的计数,未知的变异对内参和基因计数的影响是相似的.已有研究表明[20,34],这个假设在某些情况下是不可靠的.事实上,在scRNA-seq试验中添加内参仍然是极富挑战的,对内参分子添加的数量必须进行精确的校准以避免覆盖整个文库,且与内源RNA相比,外源的内参RNA分子可能会具有不同的降解和逆转录速率[26].所以,在使用基于内参的标准化方法时,应了解内参分子与所选系统的特性.
2 批次效应
“批次效应”(bach effect)通常是指当样本来自于不同批次时所产生的技术差异.例如,不同的时间,不同的操作者,不同的scRNA-seq协议,甚至不同的测序平台,都会使测序样本之间产生批次效应[35,36].理论上,可以通过设计合理的平衡试验消除批次效应.但是,因为试验操作过程中的种种限制(例如,芯片的限制,材料获取的限制等)往往无法进行标准的平衡试验.所以,为了避免由批次效应引起的技术变异而产生的误导性结论,对scRNA-seq数据进行下游分析之前,应进行批次效应校正[37,38].
已经有许多工具被开发用于消除scRNA-seq数据中的批次效应,其中包括一些原本为Bulk RNA-seq开发的方法,例如,ComBat[39]、limma[40]、DESeq2[41]等.Limma软件包中的removeBatchEffect函数可用于在进行下游分析之前删除由于批次效应和其他变异引起的系统变化.该函数通过将包含批次效应的封闭项(blocking term)线性拟合到每个基因的表达值,随后将每个封闭项的系数设置为零,使用剩余项和残差计算基因表达值,从而生成去除批次效应的表达矩阵[38,40].最近,一些专门对scRNA-seq数据进行批次效应校正的方法被提出来,例如,MNNs(projection of mutual nearest neighbors)[38]、CCA(canonical correlation analysis)[42]、f-scLVM[43]等.MNN方法主要是通过批次之间存在的MNNS消除批次效应,该方法通过MNN对之间的差异表达值进行计算,并在大量配对之后取平均值,从而提高其准确性.最后,从估计的批次效应获得校正向量,并将其运用到表达值以进行批次效应校正.但是,使用MNN进行批次效应校正有一个先决条件,即每个批次必须至少包含一个与另一个批次共享的细胞群体,否则,MNN的可靠性将会大为降低.
3 细胞类型的鉴定
获得经过标准化的数据之后,怎样高效而准确地鉴定细胞类型是scRNA-seq研究中的关键一步,通常该步骤包括降维、聚类和注释.
3.1 降维
现在主流的scRNA-seq试验可以检测出大量的基因,但同时也意味高维的数据,数据点(细胞)之间的距离变得相似,使细胞群体之间的差异与细胞内部的差异变得模糊,这被称之为“维数诅咒”[44].一般对于维数诅咒有两种处理方法,降维(dimensionality reduction)和特征选择(feature selection).降维主要是通过将高维的数据投影到低维的空间降低数据的复杂度,但是在降维的过程中不可避免地会丢失掉一些基因信息,因此降维方法的选择会涉及对特定属性优先级的划分.特征选择是指通过去掉一些无用基因,从而减少分析中的数据维度.对scRNA-seq数据聚类之前,通过这些方法降低数据的维度,有利于减少噪音并加快计算速度[45,46].
降维技术对于高维数据的可视化和下游分析至关重要,现在常用的降维方法主要分为两大类,线性降维和非线性降维.线性降维方法中最具代表性的是主成分分析(principal component analysis, PCA),PCA通过确定最大的差异方向(主要成分),将数据线性转换到由这些主要成分构成的低维空间,具有相对较快的速度,在与稀疏矩阵联用时可以在较大的数据集上使用.但是PCA因为受到线性降维和假设数据集近似符合正态分布的限制,不适合运用于scRNA-seq数据集[45].已经有一些类似于PCA的改进方法,例如ZIFA(zero-inflated actor nalysis)[47]、ZINB-WaVE(zero-inflated negative binomial model)[48],用来更好地处理scRNA-seq数据集的零膨胀问题.
非线性降维方法在scRNA-seq中的应用越来越广泛,能有效避免表征的过度拥挤,在重叠区域表示出不同的簇.最具代表性的非线性降维技术是tSNE(t-distributed stochastic neighbor embedding)[49],该技术通过为每个数据点提供二维或三维地图中的位置,实现可视化高维数据,避免将数据点集中在图像的中心以改善可视化效果,能有效揭示局部数据结构,已被运用于许多scRNA-seq数据分析.但是,tSNE具有随机性,对于同一数据集运行tSNE会产生不同的嵌入,运算速度慢,容易丢失掉长距离信息(集群间的关系),且tSNE对“困惑”(perplexity)参数敏感,往往需要多次运行,以确定合适的参数[45,50],这些特性给降维过程造成困难,需要花费大量时间选择更具生物学意义的tSNE聚类图.
其它的非线性降维方法还包括UMAP(uniform manifold approximation and projection)[51]、diffusion map(DM)[52]、Isomap[53].UMAP是最近提出的一种用于降维的流形学习(manifold learning)技术,与tSNE相比,能保留更多的全局结构,特别是子集的连续性.在运行时间上具有一定优势,且可重复性更高,是非常有发展前景的降维技术[54].但是要强调的是,在scRNA-seq数据的分析中,UMAP的实践运用仍然较少,其可靠性仍然有待检验.DM使用距离量度(通常为扩散距离)模拟细胞分化过程,并沿分化路径对细胞进行排序,同时保留数据的非线性结构[55].相比于tSNE,DM更有利于保留全局结构和伪时间顺序,但是对于较小的数据集(细胞数量少)和内部细胞差异较大的数据集,DM的表现可能不如预期[56].
特征选择往往在对scRNA-seq数据集进行预处理的时候就已经进行了,本文所介绍的许多标准化方法和批次效应校正其实就是一种特征选择.简要地说,现在常用的特征选择通常基于高度变化的基因、内参和共表达这几类方法[45].
总的来说,在scRNA-seq数据的实际分析中,往往是上述多种方法联合使用,没有哪一种方法对所有的数据类型均表现出良好的可靠性.例如,来自“液滴”体系的稀疏测序数据和Smart-seq2协议的深层测序数据,对最适软件的选择是不同的.所以,对于scRNA-seq数据的分析,应综合考虑数据特征、试验目的等因素后选择合适的分析流程.
3.2 聚类
scRNA-seq的重要用途是鉴定细胞群体的异质性,而聚类是基于scRNA-seq探索细胞异质性的关键步骤.常用的聚类工具(表1)大多数基于无监督学习算法,例如,k均值聚类(k-means)、分层聚类(hierarchical clustering)、基于密度的聚类(density-based clustering)和社区发现(community detection).

表1 常用的scRNA-seq聚类工具Table 1 Summary of scRNA-seq clustering tools
最经典的聚类算法是k-means,随机选取k个样本点作为每个簇的中心点,迭代地将样本点分配给最近的簇并重新计算簇中心.k-means算法的计算复杂度与样本点的数量呈线性关系,具有运用在大型单细胞数据集上的可能性.同时,k-means算法也存在一些局限性.例如,在运行k-means算法之前需要选定k的数值,这在某些数据集上是困难的.k-means算法更倾向于识别大小相似的簇,从而导致一些稀有的细胞种类被包含到更普遍的细胞种类中.为了克服这些问题,一些工具被开发.SC3[57]通过将不同的聚类结果合并到一个共识矩阵(consensus matrix)中,提高聚类结果的鲁棒性.RaceID[58]通过筛选背景噪音中无法解释的离群点加强对稀有细胞类型的检测,能较好地识别复杂单细胞数据集中的稀有细胞类型.但是,当数据中不包含稀有细胞种类时,RaceID的可靠性会下降[59].
分层聚类也是一种常用的聚类方法,通过对数据集在不同层次进行划分,一层一层地进行聚类.可分为两种策略,自底向上的凝聚方法(agglomerative hierarchical clustering)和自上向下的分裂方法(divisive hierarchical clustering).整个聚类过程可以视为一幅完整的树状图,能够在不同的尺度(层次)上展示数据集的聚类情况.但是,该方法运行较慢且内存占用高,对于大型数据集聚类成本高昂.CIDR[60]通过隐式插补解决单细胞数据集中的“零膨胀”问题,从而在大型单细胞数据集上实现快速的分层聚类.在分层聚类中结合降维已经成为了一种广泛应用的策略,即在每次合并或分裂后进行降维,这种迭代策略有利于对较小的簇进行识别.
基于密度的聚类方法根据样本的密度分布进行聚类,相比于k-means算法只能获得球状的聚类形状,这种聚类方法可以聚类任意的形状.由于需要大量的样本才能准确估计密度,故基于密度的聚类方法比较适合于大型单细胞数据集[45].最经典的是DBSCAN 算法[61],通常需要给定两个参数,半径(eps)和密度阈值(MinPts),类似于k-means算法中簇数k的选择.在GiniClust[62]中该算法被用于基于Gini基因表达谱的聚类,能有效识别单细胞数据集中的稀有细胞类型.在大型簇的检测中GiniClust是无效的,所以建议在使用GiniClust进行聚类的同时结合使用其他的聚类方法,避免对大型簇的检测失真.DBSCAN算法使用固定的参数进行聚类,当数据集密度不均匀时,聚类效果较差.为了解决这个问题,一些能聚类不同密度的簇的算法被提出来,例如OPTICS(ordering points to identify the clustering structure)[63]和DPCA[64].Kausar et al[65]将DPCA聚类算法引入SIMLR,替换原本的k-means聚类算法,发展出DP-SIMLR(density peaks based clustering for single-cell interpretation via multikernel learning),从而自适应的发现细胞群体中的异质性.
社区发现是越来越流行的聚类方法,专门用于基于图的数据集的聚类,是基于密度聚类的扩展.该算法对社区的大小、数量、外形无严格假设,能较好地适应非凸或非对称的社区[66].社区发现算法能在较短时间内对大型图集进行分割,意味着能较好地对大型单细胞数据集进行聚类[67].在scRNA-seq中应用最广泛的社区发现算法是Louvain算法(基于模块度优化的启发式方法)[67,68],被许多聚类工具所使用[22,69,70].Louvain算法在两个主要阶段(节点移动和网络聚合)对质量函数进行优化.在本地阶段,单个节点被移动到使质量函数增加最大的社区.在聚合阶段,基于先前的分区创建聚合网络.先前分区的每个社区将成为聚合网络的新节点,对这一过程进行迭代,直到质量函数无法优化.PhenoGraph使用最近邻图(nearest-neighbor graph)对高维的单细胞数据集进行建模,获得代表细胞之间表型关系的图.随后使用Louvain算法对图进行分割,以使模块度最大化.
值得注意的是,已经有文献报道Louvain算法在运行过程中可能会产生不良的社区连接,即被Louvain算法聚类为同一分群的细胞群内,可能存在没有关联的小分群.为了解决此问题,Traag et al[71]基于Louvain算法提出了Leiden 算法,该算法引入了智能局部移动方法,将Louvain算法中的聚合细分为分区的细化和基于细化分区的网络聚合,从而有效地保证社区之间的良好连接.
利用社区发现算法进行聚类也不可避免地具有一些限制,scRNA-seq数据缺少固有的图结构,在将scRNA-seq数据转换为图结构中常用的k邻近方法在高维数据中的可靠性可能并不鲁棒[72].事实上,尽管已经有许多聚类算法和软件被开发,但并没有一个算法和软件能适用于所有的情况.且已有研究表明[73,74],单一的算法不可能对所有类型的scRNA-seq数据集均表现出较高的鲁棒性.
3.3 注释
对所获得的簇进行注释是利用scRNA-seq进行研究的关键步骤,但是,将每个簇准确而又可靠地对应到其真实代表的细胞种类是困难的.以往研究人员通过查阅大量的相关文献和数据库鉴定每个簇的真实生物学含义.然而,直到今天,学界对于细胞类型鉴定的方法与规则仍然未达成统一的标准.
通过差异表达基因或标记基因对簇进行生物学注释是一种常用的手段.使用Gene Ontology[75]可以获得与细胞最相关的生物过程术语,这些标记基因可以用于试验验证.随着单细胞数据集数量的增长,基于先前的聚类结果对新的簇进行生物学注释成为一种可行的策略,一些“投影(合并)”工具被开发,例如,scmap[76]、MetaNeighbor[77]、Seurat3[78].scmap通过将新的簇投影到参考数据集,用质心(每个基因的表达中值向量)表示每个簇,计算新簇和参考数据集中已有簇的相似性,从而识别与新簇最为相似的簇.MetaNeighbor通过近邻元分析(meta-analysis via neighbor voting)评估细胞种类特异的转录组图谱在不同数据集之间的可复制性,并识别具有高度相似性的簇.Seurat3通过将来自于单细胞转录组、单细胞表观、单细胞蛋白质组和单细胞空间信息等不同来源的数据集“锚定”到同一共享空间,从而实现跨数据集的比较.尽管“投影”策略是优质的工具,但也仍然存在一些限制.当新簇并不存在于参考数据集时,可能会导致投影错误或无法投影[46].其次,批次效应和数据集本身的可用性都将对投影结果产生重大影响.
最近,一种介于无监督聚类和投影之间的有监督聚类策略被提出来,要求研究者提供一组标记基因列表,对单细胞数据集进行有监督聚类,从而实现对簇的自动生物学注释.Garnett[79]和CellAssign[80]是基于此策略而开发的自动注释工具.Garnett使用层次模型和分类器进行工作,CellAssign基于概率贝叶斯模型分配细胞类型.
尽管已经有各种各样的聚类和注释方法被提出来,但这些方法和工具都有着一定的限制和妥协,所以对scRNA-seq数据进行准确而鲁棒的聚类和注释仍需继续探索.也许,在不久的将来人们会对细胞类型的鉴定形成统一的规则,但在这之前,开发更加高效和准确的聚类工具仍是重点.除此之外,对由计算获得的聚类和注释信息进行试验验证也是有必要的,尽管现在的计算工具有了很大的进步,但是,试验验证仍然是更加被大家接受的金标准.
4 小结与展望
自从2009年第一次对单个细胞的转录组进行测序,scRNA-seq已经在生物学和医学领域扮演着越来越重要的角色,这项技术的发展开辟了一系列新的研究领域,给许多生物学问题带来了不同的见解.特别是在肿瘤异质性、胚胎细胞发育图谱、疾病发生机制和干细胞再生等方面的研究中发挥了重要作用,极大地提高了人们对这些与人类健康相关的生物学问题的认识.在过去的这些年,一系列scRNA-seq协议被相继提出,研究人员可以选择的空间越来越大.scRNA-seq协议的发展也给数据分析带来了巨大的挑战,单个细胞的测序成本越来越低,使研究人员能够轻易地获得数以万计的单细胞数据集.面对这些巨大、稀疏、高维的数据集,怎样无偏且准确地阐释其生物学含义,仍需要更加新颖且鲁棒的计算分析工具.
猜你喜欢
中华骨与关节外科杂志(2022年1期)2022-08-31
中国畜牧杂志(2022年8期)2022-08-13
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
水产科学(2022年1期)2022-01-26
科学导报(2021年29期)2021-06-03
教育界·上旬(2020年8期)2020-06-27
中国瓜菜(2019年8期)2019-09-19
科海故事博览·下旬刊(2019年6期)2019-04-16
江苏农业科学(2017年16期)2017-10-27
