
离子液体和低共熔溶剂催化二氧化碳合成有机碳酸酯的研究进展
2022-04-12 03:54阮佳纬叶香珠陈立芳漆志文
化工进展 2022年3期
阮佳纬,叶香珠,陈立芳,漆志文
(华东理工大学化工学院,化学工程联合国家重点实验室,上海 200237)
全球气候变暖已成为近年来的重大环境问题乃至政治问题。中国提出要在2030 年前实现碳达峰,2060年前实现碳中和,体现了中国作为负责任大国应对全球气候变化的担当,是挑战,更是机遇。二氧化碳(CO)作为主要温室气体,虽是气候变暖的要因,但也是丰富和可再生的碳一资源。基于此,国内外发展了具有应用前景的碳捕获与封存(carbon capture and storage, CCS)和碳捕获与利用(carbon capture and utilization,CCU)技术,以减缓气候变暖,同时制备高附加值化学品。CO的捕集与资源化利用已经成为能源与环境领域的重要研究方向。
目前,以CO为原料可合成有机碳酸酯、甲醇、甲烷、尿素和淀粉等多种化学品。其中,有机碳酸酯应用广泛,包括直链与环状碳酸酯。碳酸二甲酯(DMC)是一种重要的直链碳酸酯,不仅是理想的汽油和柴油添加剂,也是低毒、低黏度的绿色溶剂。环状碳酸酯在工业上可以用作极性非质子溶剂、锂电池电解质组分和有机合成中间体。在合成路线方面,CO能与甲醇直接合成DMC,与环氧化物通过环加成合成环状碳酸酯,这些符合“绿色化学”和“原子经济”的化学反应被广泛研究。
CO的热力学稳定性和化学惰性限制了其转化利用。为克服CO转化的反应能垒,人们发展了多种应用于CO合成有机碳酸酯的催化体系,其中包括金属卤化物、金属配合物、金属有机骨架和氮化碳纳米片等。但这些催化剂普遍存在反应条件苛刻、需要添加溶剂或助溶剂、成本高等缺点。离子液体(ILs)作为一种新型的绿色介质,具有结构可设计性、高催化活性和易分离特性等优势,在催化CO合成有机碳酸酯方面备受关注。低共熔溶剂(DESs)被视为新一代ILs,是由一定化学计量比的氢键受体(HBA)和氢键供体(HBD)通过氢键作用形成的低熔点混合物。它不仅具有ILs 的优良性质,还具有合成简单、价格低等优势,近年来在CO转化为有机碳酸酯反应体系中被深入研究。
本文综述了近年来国内外对于ILs和DESs催化CO转化为有机碳酸酯反应的研究进展,重点介绍了不同种类ILs和DESs的催化性能和作用机理。
1 CO2合成有机碳酸酯的反应机理
1.1 CO2与甲醇直接合成碳酸二甲酯
以CO和甲醇为原料直接合成DMC是CO高效转化途径之一。该反应受热力学平衡限制,难以实现高转化率。Kabra 等对该反应进行了热力学分析,计算了常温常压下反应的标准生成焓Δ(-16.5kJ/mol)和标准吉布斯自由能Δ(+38.0kJ/mol),表明该反应是放热反应,且在常温常压下不能自发进行;并通过实验研究发现低温高压有利于提高DMC 收率。即使在高压下,该反应平衡转化率仍不高,需要设计高效催化体系和催化脱水耦合过程。而催化剂的酸性位和碱性位对CO和甲醇的活化起重要作用,兼具酸碱活性位的ILs 在该反应中的应用逐渐得到发展。
理清CO与甲醇的反应机理有利于开发高效催化体系,文献主要结合实验与量子化学计算开展研究。Casarin 等揭示了CO在反应中既可作为电子受体也可作为电子供体,认为CO的2π(LUMO)轨道可接受来自催化剂碱性位的电子,1π(HOMO)轨道则提供电子给酸性位。因此CO在催化剂表面的活化,一种可能是通过电子从催化剂转移到CO,进而形成弯曲的CO来占据LUMO 轨道;另一种可能是电子从CO转移到催化剂表面,CO转变为CO,此时CO会保持线性构型。Tomishige 等证明了碱性位活化CO形成弯曲的阴离子,酸性位活化甲醇形成CH。Zhao 等提出了酸碱双功能ILs 催化CO和甲醇直接合成DMC 的分子活化过程和反应机理(如图1 所示):首先是碱性位作用,OH将电子转移给CO,形成弯曲的CO;甲醇在碱性的OH作用下很容易生成CHO,随后CHO亲核进攻CO中带正电荷的碳原子,形成碳酸单甲基取代阴离子CHOCOO;接着是氢键酸性位的作用,羟基和具有布朗斯特碱性的OH协同进攻甲醇,在甲醇和ILs 上的羟基间形成氢键网络,导致甲醇O—H 键和C—O 键极化,形成CH;最后,活化的CHOCOO与CH+反应生成DMC。

图1 CO2与甲醇直接合成DMC的分子活化过程与反应机理[19]
1.2 CO2与环氧化物环加成反应合成环状碳酸酯
目前工业上制备环状碳酸酯的催化剂主要是季铵盐和碱金属卤化物,由于催化活性较低,需要在高温高压下操作,每生产1吨环状碳酸酯会排放约0.9吨CO,这反而造成CO净排放。因此,开发高效催化剂尤为重要。近年来,ILs 在CO环加成反应中逐渐得到应用,多种新型ILs 在常温常压下可催化CO高效转化。为指导催化剂的开发,文献深入探讨了反应机理。
目前用于该类反应的催化剂通常包含酸碱活性位,普遍接受的ILs 催化机理是:酸性官能团(路易斯酸如金属离子等,以及HBD基团如—OH、—COOH 等)活化环氧底物,碱性官能团(卤化物、羧酸盐等)亲核进攻环氧底物上位阻较小的碳原子,随后CO被活化并插入亲核进攻中间体,最后发生分子内重排,脱除亲核基团,得到环状碳酸酯,并完成催化剂再生。
Girard 等结合实验和密度泛函理论(DFT)计算,提出了两种可能的ILs 催化CO环加成反应路径,同时探讨了潜在的副反应(如图2 所示)。在循环1中,路易斯酸中心(咪唑环2号位碳上的质子)通过与环氧化物形成氢键活化环氧底物(Ⅰ),由碱性中心卤素原子亲核进攻活化的底物,生成烷氧中间体(Ⅱ);随后CO插入烷氧中间体形成非环状碳酸盐阴离子(Ⅲ),亲核基团脱除,生成环状碳酸酯并完成催化剂再生。需要注意的是,此路径中可能发生副反应:异构化生成醛以及水合生成一元或二元醇。在循环2 中,由咪唑环-杂环卡宾结构与CO配位,形成加合物(Ⅳ);随后该加合物亲核进攻环氧底物(Ⅴ)生成烷氧基中间体(Ⅵ),最后通过分子内亲核取代得到环状碳酸酯。对循环1 进行理论计算,优化了反应物、中间产物、过渡态、最终产物的几何构型,计算了过渡态最低能量,得到总反应放热为17.35kcal/mol(1cal=4.1868J),证明了该催化反应路径的热力学可行性。

图2 CO2与环氧化物环加成反应机理[24]
2 基于离子液体的CO2转化
ILs通常指熔点低于100℃的有机盐,由有机阳离子和有机或无机阴离子组成,具有不挥发、液程范围宽、热稳定性好、溶解能力强等常规有机溶剂无法比拟的优点。在CO转化过程中,ILs 可以作为溶剂、CO吸收剂、CO活化剂、催化剂或助催化剂。文献探究了ILs 的阳离子、阴离子、烷基链长、官能团以及温度、压力、催化剂用量等对催化活性的影响。
2.1 传统离子液体
传统ILs 是一类基础ILs,包括咪唑类、吡啶类、季铵盐与季膦盐类等。国内外已经报道了大量用于催化CO转化为有机碳酸酯的传统ILs。
由于CO与甲醇直接合成DMC 反应受热力学限制,及时脱除副产物水能推动化学平衡正向移动,因此当前研究主要对高效催化剂和脱水剂进行设计。Zhao 等合成了[CCIM][HCO],在室温下将CO和甲醇合成DMC。他们发现该类ILs 会与CO形成加合物,且该加合物能结合副产物水生成咪唑碳酸氢盐,因此该ILs 可同时用作催化剂和脱水剂(如图3 所示),既突破化学平衡的限制,又简化后续的脱水剂分离,实现CO和甲醇的高效转化。Hu 等先通过咪唑环-杂环卡宾结构将CO进行化学活化,再将获得的加合物与醇直接反应,在常温常压下制备了不同的直链碳酸酯,CO转化率最高达40.2%,DMC 选择性达99.9%;结合O同位素示踪法和DFT 计算,揭示了在一卤代烃和二卤代烃存在下CO与甲醇转化为DMC 的不同反应路径,解释了卤代烃的烷基化效应以及推动化学平衡正向移动的作用。
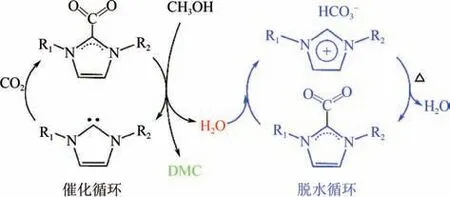
图3 咪唑碳酸氢根ILs的催化与脱水耦合机理[31]
除了移除副产物水来突破化学平衡限制,Zhang 等在尿素两步法醇解制DMC 反应中通入CO带走氨气并再生尿素以推动平衡正向移动。首先二醇基咪唑ILs 与尿素生成环状碳酸酯中间体和氨气,氨气被通入的CO带走;随后通入甲醇与环状碳酸酯进行酯交换反应,得到DMC并完成ILs的再生。在最优反应条件下,1mol尿素最多可以生成0.64mol DMC,其收率是尿素与甲醇直接反应的64倍。
传统ILs 用于CO合成环状碳酸酯也被大量报道。Anthofer 等合成了10种含不同取代基的咪唑溴盐,其中1-五氟苯基-3-正辛基咪唑溴盐在70℃、0.5MPa 下,催化环丙烷的转化率最高达到91%,碳酸丙烯酯选择性超过99%,且循环使用10次后,催化活性仍保持稳定。Xiao 等发现咪唑ILs 催化活性随咪唑环酸性的增强而增加,这是因为强酸性有利于ILs 中氮相连的质子与环氧化物的氧原子之间形成氢键来活化底物;该ILs 循环使用5 次,在保持选择性的同时,活性略有降低。Girard 等探究了一系列咪唑ILs 对CO的吸收能力,并将其用于环状碳酸酯的合成。他们发现,适量水有利于提高环状碳酸酯的收率,且无副产物二醇生成;但过量水会导致环氧底物开环和醇盐水解,加速生成副产物二醇。
Toda等报道了一种由四芳基取代的季膦盐和卤素组成的、带有布朗斯特酸性位和亲核位的ILs(如图4 所示);发现只有邻位羟基取代的季膦盐ILs 具有催化活性,且活性高,环状碳酸酯收率能达到90%,而间位和对位取代的ILs没有催化效果;还指出季膦盐中芳基上的吸电子基团会降低其催化活性。Wu等报道了一系列四丁基膦的ILs,其中[BuP][2,4-OPym-5-Ac]在常温常压下反应20h,-亚烷基环状碳酸酯收率达到91%,且对多种炔丙醇底物都具有良好催化活性。
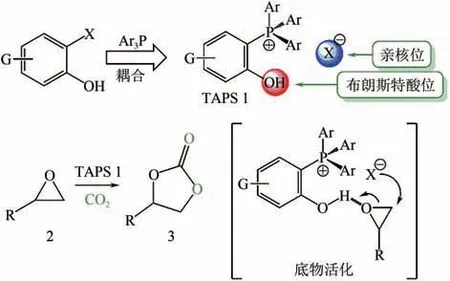
图4 四芳基卤素膦盐作为环加成反应双功能催化剂[37]
2.2 质子型离子液体
质子型ILs 是由等摩尔量的布朗斯特酸和布朗斯特碱构成的一种新型ILs。区别于其他ILs最主要的性质是其有质子从酸到碱的转移,导致存在质子供体和质子受体,因此可构建强氢键网络。此外,它还具有低成本、易制备和质子活性可调等优势,使其在CO捕集和转化方面被广泛研究。
常见的有机强碱如二甲氨基吡啶(DMAP)、1,8-二氮杂二环[5.4.0]十一碳-7-烯(DBU)等常被用作质子型ILs 的质子受体,它们能与CO形成加合物从而活化底物。Roshan 等通过DFT 计算了有机碱催化CO与环氧底物环加成过程中形成的稳定中间体和过渡态,发现CO-碱-水之间形成的HCO能提高反应活性。Meng 等制备了一系列DBU 基质子型ILs(如图5 所示),用于催化CO和环氧氯丙烷的环加成反应,其中f在常温常压下的模拟烟道气(15%CO,85%N)中反应6h,环氧氯丙烷转化率达92%;分析其傅里叶变换红外光谱发现,与CO作用后的ILs在1975cm处出现了一个对应于氨基甲酸盐的不对称振动峰,意味着该ILs的烷氧基阴离子活化了CO。理论计算表明,使用催化剂f时反应活化能最低,最有利于开环;f与环氧底物过渡态中氢键长度最短,氢键作用最强,故活化底物能力最强。需要注意的是,该ILs循环使用4次后,环状碳酸酯收率从99%降为77%,主要是环氧底物加成到ILs上导致活性位点的活性降低。
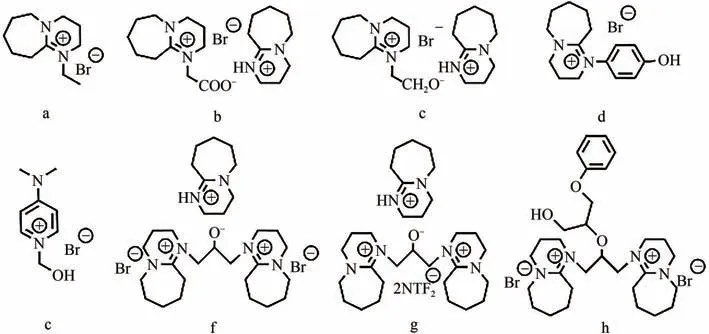
图5 质子型ILs[42]
Mujmule 等研究了有机碱DBU、三乙胺、,-二异丙基乙胺等与咪唑基ILs 组成的二元催化体系,发现DBU 碱性强且活性氮附近空间位阻较小,易于活化CO。在120℃、2MPa下,[EVIMOH][Cl]/DBU 中反应1h,环氧丙烷转化率为99.8%,产物选择性超过99%。该体系中羟基官能团、咪唑环上2号位碳上的活性质子以及含N碱性位之间的协同作用能促进CO的吸收,增强反应活性;在循环使用5 次后催化活性无明显降低。Zhang 等制备了DMAP基质子型ILs,其中[DMAPH][Br]在120℃、常压下,针对模拟烟道气反应14h,环氧苯乙烯转化率为92%。他们认为质子型ILs 的有机碱阳离子上的正电荷离域增强了与环氧化物和CO的相互作用,从而提高催化活性。该ILs 对多种环氧底物都具有良好活性,且易回收。Chen 等则通过调控ILs 的碱度,使之能同时作为吸收剂和活化剂,在常压下亚烷基碳酸酯收率达91%。
2.3 功能化离子液体
结构可调是ILs 最重要的特性。利用其结构可设计性,在阴阳离子上引入具有特殊功能的官能团可获得功能化ILs。在CO转化为有机碳酸酯的反应中,常见的功能化基团有羟基、羧基和氨基等。功能化ILs对CO表现出强吸收固定能力,在CO转化方面也表现出比常规ILs更高的催化活性。
Wang等借助DFT计算,证明了在CO与环氧底物的环加成反应中,ILs 的羟基是关键活性位。Zhao等合成了含有不同羟基数量的功能化胺基和咪唑基ILs,在催化CO和甲醇直接合成DMC 反应中,苄甲基二羟乙基氢氧化铵具有最佳催化效果,证明了醇羟基和OH的协同作用对CO的固定和活化起关键作用,另外还发现阳离子上苄基取代基带来的共轭效应有利于甲醇的羰基化反应。该ILs 在循环使用4次后,甲醇转化率有明显降低,可能是由于回收时的质量损失。Sun 等开发了羟基功能化ILs,在[HEMIM][Br]中,125℃、2MPa 下反应1h,环氧丙烷转化率高达99.2%,碳酸丙烯酯选择性达到99.8%。
Sun等还开发了羧基功能化的咪唑ILs用于催化CO与环丙烷的环加成反应,考察了氢键强弱和酸度对催化活性的影响,认为酸碱双功能ILs 催化剂的强酸性能增强阳离子的开环能力,但也会削弱阴离子的亲核性,最终降低碳酸丙烯酯产率。其循环使用5次后,催化活性无明显下降,转化率仍达97%,选择性为99%。Xiao 等发现弱酸有利于提高催化活性,酸性过强时会形成强氢键而阻碍CO的插入,降低目标产物产率。Meng 等发现可以通过控制羧酸基ILs 的烷基链长来调节其在反应体系中的溶解度。在加热条件下形成均相,从而表现出最大催化效率;反应结束后降温,ILs 与产物分相,易于分离。
氨基功能化咪唑ILs 由于其活化和固定CO的能力,能有效催化CO与环氧化物的环加成反应。Chen等通过DFT计算指出,氨基功能化咪唑ILs 中咪唑环主要催化开环,质子化的胺基则稳定溴离子的亲核进攻。Yue等在[APBIM][I]中,在120℃、1.5MPa 下反应1.5h,碳酸丙烯酯收率能达94.3%。他们指出,阳离子上较长的烷基链有助于提高ILs 的催化活性,而卤素阴离子的催化活性顺序为I>Br>Cl,这与Liu 等得到的结果一致。此外,氨基功能化ILs 既能与CO反应生成氨基甲酸盐来活化CO,又能通过氨基上的质子与环氧底物的氧原子形成氢键来活化环氧底物,从而实现对CO和底物的双重活化。Yue 等尝试了不同种氨基酸作为阴离子的咪唑基双功能ILs 在90℃、0.25MPa 下催化CO与环氧底物的环加成反应,反应12h后碳酸环氯丙烯酯最高收率达到99%。该氨基酸ILs在200℃下不会分解,循环使用4次后,催化活性略有降低。
传统ILs 稳定性较好,但反应条件较苛刻,催化活性也偏低。质子型ILs 易制备、催化活性高,近年来已有在常温常压下仍具有高催化活性的质子型ILs 被报道,但其稳定性仍需提高。多种功能化ILs 能获得超过95%的产物收率,但反应条件较苛刻,难以在温和条件下催化转化。此外,多数功能化离子液体黏度较大,工业化应用有所局限。
3 基于低共熔溶剂的CO2转化
ILs因制备复杂、难生物降解、成本高等缺陷,不利于工业化应用。DESs是HBA与HBD在一定比例下通过氢键作用结合形成的低熔点混合物。常见的HBA 包括氯化胆碱、季铵盐、季膦盐等,HBD 包括醇、羧酸、酰胺、糖类等。DESs 具有廉价、易获取、可生物降解、无毒等ILs不具备的优良性质,被认为是ILs 的潜在替代品,适合规模化生产。
DESs 的独特理化性质使之在气体吸收、液体分离、反应催化、电化学等领域表现出优异性能。在CO捕集方面,报道较多。Fu 等制备了DBU 与吡咯组成的DESs,发现实际吸收量远大于理论吸收量,并证明DESs 解离产生的离子化程度决定其吸收能力。此外,有研究指出DESs 对CO的吸收能力与其酸度或碱度无关,而与其HBA 和HBD之间的相互作用强弱有关。
在DESs 用于CO化学转化方面的研究尚处于起步阶段。通过改变HBA 与HBD 的种类和配比,可以调控对特定反应物的氢键形成能力,提高DESs 的吸收能力与催化活性。目前,以氯化胆碱、季铵盐与季膦盐为HBA 构成DESs 催化CO与环氧底物反应的报道较多,有机碱和其他类型的HBA 较少。本文涉及DESs 的组成如图6所示。
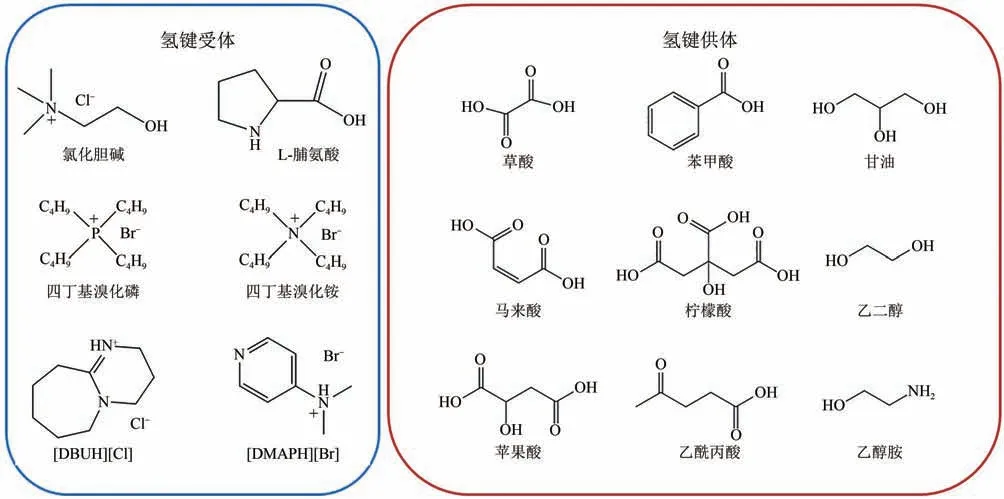
图6 本文所涉及DESs的组成
3.1 氯化胆碱基低共熔溶剂
氯化胆碱无毒、生物相容性好、价格低廉,是组成DESs最常用的HBA。由其构成的DESs制备简单,可生物降解,而且可以通过改变HBD 种类与配比来改变DESs的物理化学性质。
Tak等首次报道了在DESs中将螺环氧吲哚通过环加成合成新型螺环碳酸酯的策略,并以甘油、乙二醇、苯磺酸和尿素作为HBD 制备了氯化胆碱基DESs。在70℃、常压下经过2h,在氯化胆碱/尿素组成的DESs催化下螺环碳酸酯产率能达到98%。Cheng等首次将氯化胆碱和ZnBr组成的路易斯酸性DESs用于CO环加成反应,在110℃、1.5MPa下反应1h,环状碳酸酯收率高达99%,TOF 为494h。Liu 等开发了基于-羟基丁二酰亚胺的DESs,在碘化胆碱与-羟基丁二酰亚胺以1∶2摩尔比组成时,在室温下反应10h,环氧丙烷转化率达到96%。该DESs 可以通过萃取分离,且循环使用5 次催化活性无明显降低。Dindarloo Inaloo 等制备了无水合金属氯化物和氯化胆碱组成的DESs,将CO、胺和卤代烃三组分偶联成氨基甲酸酯,发现氯化胆碱和氯化锌以1∶2的摩尔比组成的DESs具有最好的催化活性,室温下反应2h 产物收率达95%。该DESs 循环使用6 次后催化活性略有降低,多种底物仍能获得90%以上的氨基甲酸酯收率。
氯化胆碱还能与生物质衍生的有机酸和醇形成DESs。Vagnoni 等将乙二醇、甘油等醇类HBD 和草酸、柠檬酸、苹果酸、马来酸、酒石酸等羧酸类HBD与氯化胆碱、碘化胆碱等HBA制备的DESs用于CO与环氧底物环加成反应,发现碘化胆碱基DESs 在常压下的催化活性普遍高于氯化胆碱基DESs,这归因于I优秀的离去能力;HBA 和HBD在未形成DESs时的催化能力较形成DESs后有明显下降,说明DESs 具有更完整的氢键网络,活化底物能力更强。
3.2 季铵盐和季膦盐基低共熔溶剂
季铵盐是工业上合成环状碳酸酯常用的催化剂,由于催化活性低,需要对其进行优化。而形成DESs 是一种可行方式,既避免使用有机溶剂,又可以利用HBA与HBD的协同作用提高催化活性。
Wang 等开发了以脂肪族羧酸作为HBD,季铵盐作为HBA 的DESs,在80℃、0.4MPa 下,环氧丙烷转化率最高达98%,碳酸丙烯酯选择性超过99%;发现pa值为3.7~4.9的脂肪族羧酸与四丁基溴化铵组成的DESs 催化活性最佳,底物转化率均大于95%。Yingcharoen 等以不同pa 值的酚、一元和多元醇、羧酸、抗坏血酸等作为HBD,四丁基溴化铵、四丁基碘化铵等作为HBA,合成了DESs,考察了在CO与环氧氯丙烷的环加成反应中前20 min的初始反应速率常数,发现pa值为9~10.5的HBD 所形成的DESs 催化活性最高。Wang 等合成了不同羟基取代位置的吡啶和四丁基铵盐组成的DESs,其中3-羟基吡啶与四丁基碘化铵形成的DESs 反应速率最快,环状碳酸酯收率也最高,能达到95%。Liu 等将苯酚,苯胺,苯磺酸和邻、间、对位氨基苯酚作为HBD,与四丁基溴化膦形成DESs,用于环氧底物环加成反应,在四丁基溴化膦与间氨基苯酚以1∶2摩尔比组成的DESs催化活性最高,其循环使用5 次,催化活性无明显降低。Liu 等以四丁基溴化胺为HBA,与乙二醇、1,2-丙二醇、三甘醇、丙二酸、葵酸等HBD 制备了一系列DESs,用于环氧化大豆酸甲酯与CO的环加成反应,在最优化条件下反应10h,大豆酸甲酯收率达到95%。
季铵盐与季膦盐作为HBA,除了能与羧酸、醇类等形成DESs,也能与金属卤化物作用形成DESs。Sun等发现在ZnCl/PPhCHBr(摩尔比1/6)中,在120℃、1.5MPa下反应1h,环氧丙烷转化率达到96%,TOF达4718.4h。Rehman等发现在季铵盐与ZnBr组成的DESs体系中,卤代季铵盐中阴离子活性顺序为Br>I>Cl>F。Br由于其优秀的亲核能力和离去能力,表现出最高的催化活性。
3.3 有机碱基低共熔溶剂
有机碱作为HBA形成的DESs与有机碱基质子型ILs类似,组成DESs的HBD可以是布朗斯特酸,也可以是醇、醇胺等。此外还可以将有机碱基ILs与HBD以一定的摩尔比混合,得到DESs。
Sun 等研究了以有机碱DBU 和纤维素组成的DESs 催化CO与环氧底物的环加成反应,在120℃、2.0MPa 下反应2h 环氧丙烷转化率为93%。对比实验及原位红外光谱,推断DBU 和纤维素对CO与环氧底物的活化具有协同作用,并提出了可能的催化机理(如图7 所示),纤维素活化底物,DBU 既进攻环氧底物,又与CO形成加合物活化CO,两个中间体偶联后DBU与纤维素脱除并完成再生,得到环状碳酸酯。
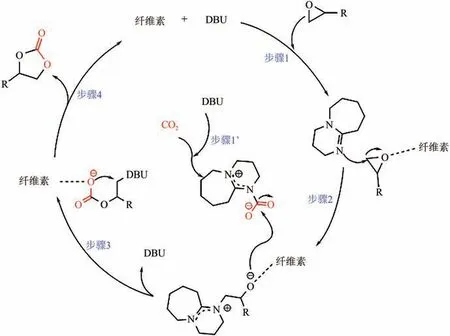
图7 DBU-纤维素体系催化CO2环加成反应的作用机理[80]
Garcia-Arguelles 等以超强碱1,5,7-三叠氮双环[4.4.0]癸-5-烯(TBD)和DBU为HBA,苯甲醇、乙二醇、甲基二乙醇胺等醇类为HBD,合成了DESs。发现提高HBD 比例可以提高对CO的吸收能力,而1∶1摩尔比的TBD与苯甲醇形成的DESs在100℃、1.2MPa 反应条件下,CO环加成反应中环状碳酸酯收率最高达到98%。Yang等将质子型ILs 作为HBA,与尿素、乙醇胺、二乙醇胺(DEA)、甲基乙醇胺等HBD 形成DESs,在常温常压下,[DBUH][Br]-DEA(2∶1)中碳酸苯乙烯酯收率能达97%。
3.4 其他类型
除了上述组成,文献还报道了氨基酸等其他类型的HBA 组成的DESs。Lyu 等以氨基酸为HBA,与二元酸合成了多种天然DESs,在130℃、1.2MPa下反应5h,碳酸丙烯酯收率高达98.6%。值得注意的是,这类DESs需要在共催化剂ZnBr的协助下才能实现高收率。Wang 等开发了由[BMIM][Cl]/硼酸/戊二酸组成的三元DESs 并将其用于催化CO与环氧丙烷的环加成反应,在配比为7∶1∶1时,碳酸丙烯酯收率最高能达98%。该三元DESs 催化体系循环使用5次,选择性保持不变,但由于产物覆盖了活性位点以及分离过程中DESs 的损失,底物转化率有所降低。
DESs 在CO捕集与催化转化方面已取得一定研究进展,但仍存在稳定性较差、后续分离困难等问题。DESs 作为二元或多元混合物体系,由于HBA 与HBD 的沸点差异,与有机溶剂和ILs 相比,在复杂体系中其分离更困难,用传统的分离方法回收的DESs 可能因其损失难以恢复到原始组成。开发能获得高纯产品的催化剂和过程工艺,将更具有学术和应用价值。
4 结语
目前ILs 在CO与甲醇合成DMC、与环氧化物合成环状碳酸酯反应中已获得较高催化活性,但也存在反应条件苛刻、成本高、分离困难等缺点。高效ILs 催化剂通常具有以下结构:①兼具酸性位与碱性位,酸碱活性位能有效活化反应底物和CO,调控阴阳离子酸碱度能实现对不同反应体系的高效催化活性;②阴离子通常选取兼具强亲核性和强离去能力的基团,如卤素阴离子;③阳离子通常选取体积大、具有电子离域作用的基团以及大分子有机碱,如咪唑、吡啶、DBU、DMAP等常作为阳离子。
DESs被视为ILs的廉价替代品,对其研究虽起步较晚但发展迅猛。DESs 在催化DMC 合成中的研究还很少,在环状碳酸酯合成中DESs 催化剂尚存在催化活性不高、反应底物种类较少等不足。在后续研究中,可以重点关注以下问题。
(1)DESs 在有机碳酸酯合成反应中的催化机理尚需深入研究,理清HBA 和HBD 与反应体系之间的分子相互作用有助于解释DESs 结构与性能之间的关系。
(2)探索更多种类的HBA和HBD。根据ILs开发的经验,开发基于多尺度计算或深度学习的计算机辅助设计方法,有助于针对特定反应体系筛选或设计出合适的DESs。
(3)开发能在温和条件下,催化低浓度CO与甲醇或来自可再生资源的生物基环氧化物转化为有机碳酸酯的DESs,更有利于环境与经济可持续发展。
猜你喜欢
教育周报·教育论坛(2020年3期)2020-10-21
中国建筑金属结构(2019年4期)2019-05-15
科技资讯(2018年16期)2018-10-26
科技信息·下旬刊(2018年8期)2018-10-21
哈尔滨理工大学学报(2018年6期)2018-02-13
新高考·高二数学(2016年11期)2017-07-06
科技资讯(2017年12期)2017-06-09
科技创新与应用(2017年9期)2017-04-26
科技视界(2016年23期)2016-11-04
医学美学美容·中旬刊(2015年2期)2015-10-21
