
两种流动相系统对氟康唑原料药含量测定的方法学对比研究
2022-04-11 13:15梁艳辉邢征宇姚传华
广州化工 2022年6期
梁艳辉,邢征宇,刘 斌,姚传华,魏 丹
(海南省食品药品检验所海口分所,海南 海口 570100)
氟康唑为三唑类第三代抗真菌药,被用于各种真菌感染特别是对隐球菌、念珠菌和皮肤真菌具有良好的抗菌作用[1-2]。《中国药典》2020年版二部收载的氟康唑原料药的含量测定为电位滴定法,该方法操作繁琐[3]。本文参考《中国药典》2020年版二部氟康唑原料药有关物质项下检测方法和其制剂项下含量测定方法选择了两种系统对氟康唑原料药的含量进行了方法学研究。
1 仪器与试药
1.1 仪 器
Agilent 1260型高效液相色谱仪,美国安捷伦公司;XS205DU 型电子天平,梅特勒-托利多(上海)有限公司。
1.2 试 药
氟康唑原料药(批号:210902),海南沙汀宁制药有限公司;氟康唑对照品(批号:100314-201906,含量:99.8%),中国食品药品检定研究院;氟康唑杂质Ⅰ(批号:130699-201501,含量:99.1%),中国食品药品检定研究院;甲醇和乙腈均为色谱纯,其它试剂均为分析纯,水为超纯水。
2 方法与结果
2.1 高效液相色谱法
在系统1和系统2的色谱条件相同的部分为色谱柱均为ACE Excel 5 C18(4.6 mm×250 mm,5 μm),流速均为1.0 mL·min-1,进样量均为20 μL,柱温均为40 ℃,方法系统适用性要求为氟康唑与杂质Ⅰ的分离度大于1.5,理论塔板数大于2000。流动相比例和实验结果系统使用性见表格1。

表1 高效液相色谱法中色谱条件
2.2 溶液的配制
对照品溶液:精密称取氟康唑对照品约50 mg+分别用系统1和系统2流动相→50 mL。
供试品液溶液:精密称取氟康唑样品约50 mg+分别用系统1和系统2流动相→50 mL。
杂质Ⅰ对照品溶液配制:精密称取杂质Ⅰ对照品约10mg+分别用系统1和系统2流动相→10 mL。
系统适用性溶液的配制:精密称取氟康唑10 mg+分别用系统1和系统2流动相+精密量取杂质Ⅰ对照品溶液1 mL→10 mL。
2.3 方法学验证
2.3.1 专属性试验
精密称取氟康唑样品约50 mg,置50 mL容量瓶中,加系统1和系统2流动相溶解后分别做如下处理:(1)酸破坏:加 1 mol/L盐酸溶液0.2 mL,放置1 h,再加1 mol/L氢氧化钠溶液0.2 mL中和,再分别用系统1和系统2流动相稀释至刻度;(2)碱破坏:加入1 mol/L氢氧化钠溶液0.2 mL,放置1 h,再加1 mol/L盐酸溶液0.2 mL中和,再分别用系统1和系统2流动相;(3)氧化破坏:加入30%过氧化氢溶液0.2 mL,放置 1 h,再分别用系统1和系统2流动相溶解并稀释定容至刻度;(4)高温破坏:取本品置80 ℃烘箱放置3 h,精密称取50 mg,置50 mL量瓶中,再分别用系统1和系统2流动相溶解并稀释定容至刻度,摇匀。(5)光照破坏:取本品光照破坏24 h,精密称取50 mg,置50 mL量瓶中,再分别用系统1和系统2流动相溶解并稀释定容至刻度,摇匀。按2.1.1项下的色谱条件测定,详见图1。结果表明,原料药在酸、碱、光和热的破坏试验中,未出现其他杂质。在氧化破坏的条件下产生较多降解杂质,各杂质峰与主成分峰的分离度均大于1.5,表明本法的专属性良好。在将原料药制备成制剂时,应重点关注其在氧化环境中产生的杂质会影响到制剂的稳定性,预知未知杂质,则需要进一步研究。

图1 系统1和系统2高效液相色谱破坏图
2.3.2 线性范围试验
系统1中氟康唑对照品溶液进样量在3.1048~31.0478 μg范围内,进样量与峰面积呈良好的线性关系,线性方程为Y=116.9X+7.765,r=1;系统2中氟康唑对照品溶液进样量在2.0958~31.4370 μg范围内,进样量与峰面积呈良好的线性关系,线性方程为Y=115.15X+5.61,r=1。
2.3.3 重复性和精密度试验
精密称取氟康唑6份样品,按照2.2要求稀释后,平行测定6次,系统1中6份样品的平均含量为99.7%,RSD为0.3%(n=6);系统2中6份样品的平均含量为99.4%,RSD为0.2%(n=6),结果表明氟康唑样品在系统1和系统2中均具有良好的重复性和精密度。
2.3.4 稳定性试验
供试品溶液在系统1和系统2中连续12小时进样检测,样品含量RSD分别为0.05%和0.09%,表明样品在12小时内稳定性较好,能够满足检验要求,结果见表2。

表2 系统1和系统2稳定性试验结果
2.3.5 检测限与定量限
将对照品溶液逐级进行稀释,测得其检测限(S/N=3)和定量限(S/N=10),系统1中的检测限为2.0958 ng,定量限为8.3832 ng;系统2中的检测限为0.0414 ng,定量限为1.0349 ng。
2.3.6 耐用性试验
在系统1和系统2中其他条件不变的情况下分别考察了氟康唑6份样品在调整流速和柱温发生变化时,其含量的变化情况。考虑到系统误差和人为误差后,流速和柱温发生变化后,两两比较的RSD值均在合理的范围内,具体实验过程中可以根据实验室的条件选择合适的实验方案,结果分别见表3和表4。
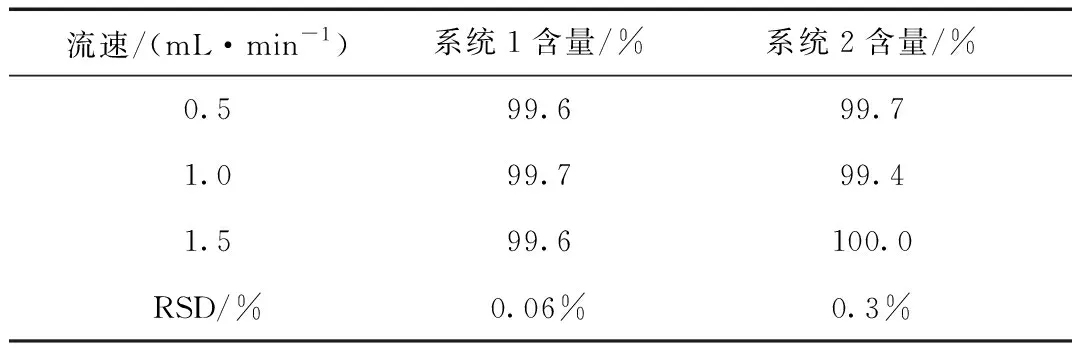
表3 系统1和系统2不同流速对含量测定的影响

表4 系统1和系统2不同柱温对含量测定的影响
3 结 论
本实验考察了两种系统对氟康唑的影响,实验表明两种流动相系统对含量检测结果影响相差不大,系统适用性试验结果表明系统1的分离度和理论塔板数优于系统2。《中国药典》2020年版二部中,系统1是原料药的有关物质检测系统,系统2则用于其制剂中含量测定和有关物质。试验对方法的专属性进行全面考察,通过破坏实验结果表明,降解杂质与主成分分离度均符合要求,王成刚[4]报道了在专属性考察中保留时间约为17.5 min的杂质是由生产工艺过程中产生;严小红[5]报道了氟康唑中未知杂质均在主峰相对保留时间1.5以上,均能与主峰获得良好分离,对含量测定结果无干扰。耐用性试验表明合理范围内的条件参数变化,对结果影响较小,表明方法耐用性良好。
原料药的纯度要求高,限度要求严格,一般为99.0%~102.0%,含量测定更加注重结果的准确性。《中国药典》2020年版对氟康唑原料药的含量测定采用的是非水溶液滴定法,随着现代药品检测技术的提高,高效液相色谱法既可用于定性分析又可用于定量分析,检测结果准确度灵敏度均较高,在药品研发、质量控制和检测等领域有着广泛的应用。方法验证试验表明,两种系统均可用于氟康唑原料药的含量测定,且可优选系统1的方法。
猜你喜欢
化学工程师(2022年9期)2022-10-21
健康体检与管理(2022年4期)2022-05-13
中国经济周刊(2021年22期)2021-12-07
艺术品鉴(2020年6期)2020-12-06
中华养生保健(2020年5期)2020-11-16
环球时报(2020-02-20)2020-02-20
传媒评论(2019年6期)2019-10-14
中国抗生素杂志(2019年1期)2019-01-30
中国经济信息(2017年17期)2017-09-09
领导文萃(2017年6期)2017-03-24
