
Phylogenetic, evolutionary, and biogeographic origin of the genus Sheathia (Batrachospermales, Rhodophyta)*
2022-04-07 09:09JinfenHANFangruNANJiaFENGJunpingQiLIUXudongLIUShulianXIE
Jinfen HAN, Fangru NAN, Jia FENG, Junping LÜ, Qi LIU, Xudong LIU, Shulian XIE
School of Life Science, Shanxi Key Laboratory for Research and Development of Regional Plants, Shanxi University, Taiyuan 030006, China
Abstract The genus Sheathia consists of over 20 species primarily distributed in Asia, Europe,Oceania, North America, and Africa. However, the origin and evolution of this genus remain unclear.Two gametophyte stage specimens (SAS18052 and SAS18523) and two “Chantransia” stage specimens(YTS19161 and YTS19017) were collected from Shanxi and Henan Provinces in China, respectively.Based on morphological data, isolates YTS19161 and YTS19017 were similar to Audouinella pygmaea,whereas the morphological characteristics of SAS18052 and SAS18523 were in good agreement with the circumscription description of S. longipedicellata. Molecular sequences of rbc L, COI-5P, and psb A were used to investigate the phylogenetic, evolutionary, and biogeographic origin of the genus Sheathia. The three molecular markers supported that the two gametophyte stage specimens belong to S. longipedicellata,while the isolates YTS19161 and YTS19017 were the “Chantransia” of S. longipedicellata. Ancestral area reconstruction and divergence time estimation speculated that Sheathia originated in North America, a portion of the Pangaea at approximately 328.07-184.73 million years ago (Ma). Our relaxed molecular clock analysis suggests that the Florideophyceae diverged approximately 741.04 (894.36-631.70) Ma. The major divergences in this class involved the emergence of Nemaliophycidae [ca. 662.01 (779.83-580.51)Ma], and the split of orders Batrachospermales and Thoreales [ca. 456.10 (552.80-367.88) Ma].
Keyword: divergence time; geographic origin; molecular analysis; morphology; phylogenetic relationship
1 INTRODUCTION
SheathiaSalomaki & M. L. Vis 2014, belonging to Rhodophyta, Florideophyceae, Batrachospermales,and Batrachospermaceae, is a typical Rhodophyta algal genus that inhabits exclusively in streams or rivers (Salomaki et al., 2014). Most of the Florideophyceae taxa have a triphasic life history,including gametophyte, carposporophyte, and tetrasporophyte. However, in freshwater red algae,except for a few species alternating with three typical generations, most species, including Batrachospermales taxa, alternate between gametophyte and carposporophyte. Nevertheless, a diminutive diploid degenerated tetrasporophyte“Chantransia” stage exists between gametophyte and carposporophyte in the life history of the Batrachospermales order (van den Hoek et al., 1995;Han, 2012; Johnston et al., 2018). Like other taxa of the Batrachospermales order, theSheathiagenus prefers to live in low temperature, low light, high dissolved oxygen, and clean springs and streams. This genus is characterized as gelatinous gametophyte f ilaments, with a beaded appearance; the whorls are spherical, and either separated or adjacent; the undiff erentiated carpogonial branches typically arise from periaxial cells, with a small number of unstalked carpogonia occurring on periaxial cells; the carposporophytes are spherical within or stretching out of the whorls (Salomaki et al., 2014). Furthermore,the most typical feature of this genus is heterocortication. However, this feature is absent in several species of this genus, includingS.absconditaStancheva, Sheath & M. L. Vis;S.arcuata(Kylin)Salomaki & M. L. Vis;S.assamicaNecchi, J. A.West, E. K. Ganesan & Yasmin;S.californicaStancheva Sheath & M. L. Vis;S.dispersaNecchi, J.A. West, E. K. Ganesan & S. K. Rai;S.indonepalensisNecchi, J. A. West, E. K. Ganesan, S. K. Rai & F.Yasmin;S.longipedicellata(D. Hua & Z. X. Shi) J.-F.Han & al.;S.murpheyiA. L. Szinte, J. C. Taylor & M.L. Vis;S.plantuloidesM. L. Vis; andS.transpacif icM. L. Vis (Necchi et al., 2019c; Vis et al., 2020).
BatrachospermumlongipedicellatumS. L. Xie &J. Feng (currently regarded asS.longipedicellata)was previously reported in China’s Jiangsu Province.It is characterized by spherical and separated whorls of 600-1 000 μm in diameter, dense and irregular fascicles arising from internode, carpogonial branches of 5-9 cells arising from primary fascicles or cortical f ilaments, 23.0-35.0-μm long and 4.5-6.0 μm in diameter carpogonia, spherical carposporophytes stretching out of the whorls, and 10.0-11.0-μm long,8.0-9.0 μm in diameter carposporoangia (Hua and Shi, 1996). Originally,B.longipedicellatumwas classif ied into the sectionBatrachospermumof the genusBatrachospermum. Until 2014, this species was transferred into the sect.Helminthoideaof genusBatrachospermumbased oncox2-3 spacer sequences(Ji et al., 2014). Subsequently, Han et al. (2018)transferred this species to the genusSheathiabased on the large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase (rbcL) and the photosystem II reaction center protein D1 (psbA) genes.Sheathialongipedicellatais endemic to China. According to Sun et al. (2001),S.longipedicellatais more suitable for growing in springs with low and constant water temperature, pH 7.5, high calcium content, and low nutrient elements content.
Molecular phylogenetic analyses based onrbcL,psbA, and the 5’ region of the mitochondrial cytochrome c oxidase I gene (COI-5P) have been widely used in phylogenetic inference and phylogenetic position examination of freshwater red algae. DNA sequence data for therbcL and COI-5P have recently become available for solving the paraphyletic of genusBatrachospermum. Based on the molecular evidence, several sections, includingVirescentia,Helminthoidea,Turfosa,Acarposporophytum,Aristata, andMacrosporasections have been raised to the genus level (Salomaki et al., 2014; Necchi et al., 2018, 2019a, b; Vis et al.2020). The f irst freshwater coralline alga was reported based on molecular phylogenetic analysis of thepsbA gene (Žuljević et al., 2016). Furthermore,rbcL andpsbA have been analyzed to determine the affi nities of four putative “Chantransia” isolates collected from China, and two new genusSheathiaspecies have been proposed (Han et al., 2020).
Sheathiais a typical genus of the Rhodophyta phylum. To date, species attributable to this genus have been reported from numerous locations in Asia,Europe, Oceania, North America, and Africa (Xie and Ling, 2004; Shi, 2006; Salomaki et al., 2014; Han et al., 2020). The research on the ancestral geographical origin and divergence time estimation of the genusSheathiais fundamental in describing the evolutionary history of the Rhodophyta taxa. However, although various investigations on the divergence time and historical biogeography of Rhodophyta based on genome and individual genes have been reported recently (Feng et al., 2015; Yang et al., 2016; Nan,2017), no related reports have covered members of the genusSheathia.
Based on the morphological and molecular data,the phylogenetic position of the fourSheathiataxa,including both gametophyte and “Chantransia”stages, was determined. Furthermore, based on the modern geographical distribution of theSheathiagenus, a preliminary analysis of the origin and ancestral geographical distribution of the genus was performed. To further clarify the evolution of genusSheathia, divergence times were estimated using Bayesian relaxed-clock methods.
2 MATERIAL AND METHOD
2.1 Sample collection and morphological observation
FourSheathiasamples were collected from Shanxi and Henan provinces in China. Samples included gametophyte (SAS18052 and SAS18523) and“Chantransia” (YTS19161 and YTS19017) stages.
Table 1 shows collection information. Materials were rinsed several times in ultrapure water, removing impurities and epiphytic algae.
After a preliminary morphological observation using a BX-51 Olympus microscope equipped with a charge-coupled device (DP72; Olympus, Tokyo,Japan), samples were divided for morphological and genetic analyses. For morphological analysis each sample was f ixed in 4% formalin solution. For DNA extraction, samples were stored in silica gel. Voucher specimens were deposited in the herbarium of Shanxi University (SXU). Voucher information is detailed in Table 1.

Table 1 Sample information for taxa analyzed in this study
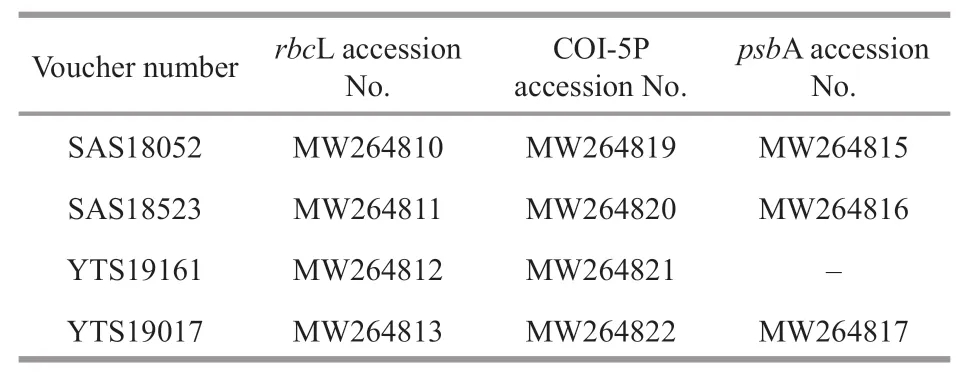
Table 2 GenBank accession numbers of rbc L, COI-5P, and psb A sequences generated in this study
Critical diagnostic morphological features (Necchi and Zucchi, 1995; Kumano, 2002; Salomaki et al.,2014) of each sample were observed via a BX-51 Olympus microscope equipped with a charge-coupled device.
2.2 DNA extraction, amplif ication, and sequencing
Total DNA was extracted following the protocol originally described by Saunders (1993) and revised by Vis and Sheath (1997). Three molecular markers,includingrbcL, COI-5P, andpsbA, were amplif ied using the primers and protocols described by Vis et al.(1998) and Saunders (2005). PCR products sequencing was performed as described previously in Han et al.(2018, 2020).
2.3 Phylogenetic analyses
Sequences generated in this study were submitted to the GenBank databases (Table 2). Additional related sequence data of the Batrachospermales order and outgroup taxaBangiawere downloaded from GenBank (http://www.ncbi.nlm.nih.gov/) (listed in Supplementary Table S1). The 65rbcL, 47 COI-5P,and 42psbA sequences were aligned by Clustal-X 2.0(Thompson et al., 1997) and MEGA 5.0 (Tamura et al., 2011). Pairwise distance and the number of nucleotide variances for the taxa’s molecular markers were calculated via MEGA 5.0. For phylogenetic analyses, the appropriate models for the sequence evolution were determined by Modeltest 3.7 (Posada and Buckley, 2004) (Table 3). PHYML software(Felsenstein, 1981; Guindon and Gascuel, 2003) was utilized to construct the Maximum Likelihood (ML)trees with 1 000 replicates of bootstrap analysis.Bayesian Inferences (BI) were performed in MrBayes version 3.1.2 (Ronquist and Huelsenbeck, 2003) and runs 5 000 000 generations sampling every 1 000 generations until the standard error was lower than 0.01. The resulting phylogenetic trees were edited using Figtree1.3.1 (http://tree.bio.ed.ac.uk/software/f igtree/).
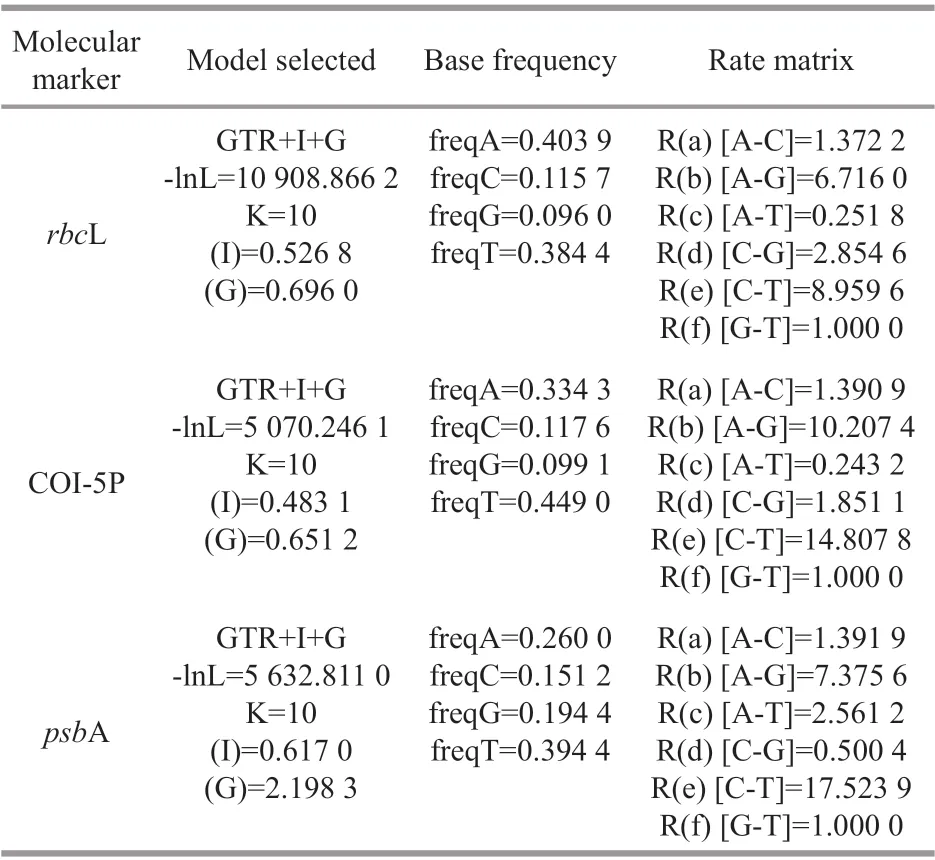
Table 3 Substitution models obtained for each gene sequence using Modeltest 3.7 analysis
2.4 Estimation of divergence time
We estimated the divergence time of clades on combined sequences (rbcL, COI-5P, andpsbA)matrix using Beast v1.7.5 (Drummond and Rambaut,2007). An uncorrelated lognormal relaxed clock model was used to account for the uncertainty in the divergence time estimation (Drummond et al., 2006).To estimate the divergence time for each node, eight fossil calibrations were used to constraint time. Four fossil calibrations were used for divergence time estimation of Rhodophyta. The f irst calibration was the 1 047 (+13/-17) million years ago (Ma)Bangiomorphapubescensfossil in the Bylot Supergroup of Baffi n Island, which was used to constraint the divergence between rhodophytes and viridiplantae (Gibson et al., 2018). The secondcalibration was the Corellinalean algae from the Doushantuo Formation, which can be traced back to 635-551 Ma and belonged to the modern red algal subclass Corallinophycidae (Xiao et al., 1998;Condon et al., 2005). Two calibration points in Corallinophycidae were the 136-130 Ma for the Sporolithales split and 120-114 Ma for Corallinales and Hapalidiales split (Aguirre et al., 2000, 2010).Additionally, four calibration nodes were used to constraint green algae and plants based on three fossil records, including 480-471 Ma for the age of land plants, 422-401 Ma for the age of the Euphyllophytes,351-313 Ma for the age of the seed plants, and 163-138 Ma for the split of Eudicotyledoneae (Magallón et al., 2013). The program Tracer v 1.6 (Rambaut et al., 2014) was used to analyze the results. When the adequate sample size (ESS) for all relevant parameters was above 200, it was assumed that stationarity had been reached. Finally, the f inal tree was viewed and edited in FigTree 1.3.1 (Rambaut, 2009).
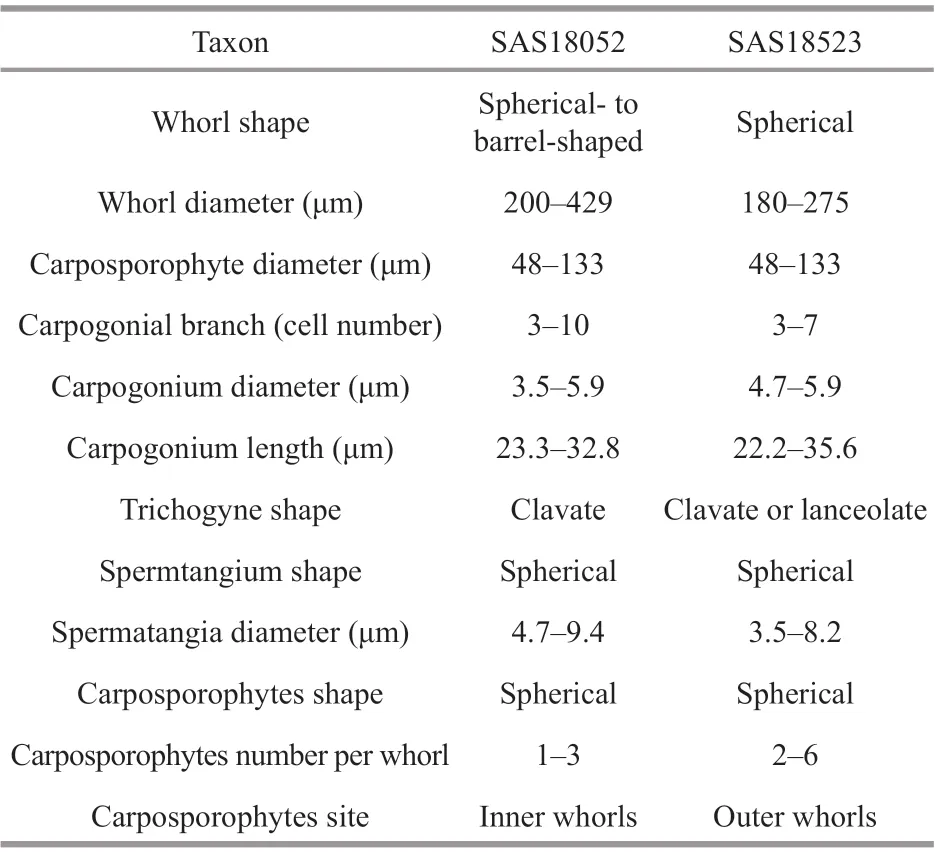
Table 4 Morphological characteristics of gametophyte stage specimens in this study
2.5 Ancestral geographical origin of the genus
rbcL and COI-5P gene sequences ofSheathiataxa and other species distributed in diff erent areas were used to reconstruct the ancestral area (Supplementary Fig.S1, Supplementary Table S2). Ancestral area reconstructions ofSheathiawere inferred using Reconstruct Ancestral State in Phylogenies (RASP)based on the tree data and the f inal tree generated in Beast. Ancestral areas at the internal nodes within the phylogenetic tree were inferred using the Bayesian binary method (BBM) implemented in the RASPsoftware (Nylander et al., 2008). Distribution ranges forSheathiaand other taxa of orders Batrachospermales and Thoreales were divided into six areas: A, Asia; B,Europe; C, Oceania; D, North America; E, South America; and F, Africa. During the Carboniferous to Triassic period, the continental plates collided with each other, causing all the land in the world to be closed together, forming the Pangaea supercontinent(Scotese et al., 1979). Therefore, the maximum areas of each node were set to 6, and the geographical combinations were set following this def inition. All other options remained as default.
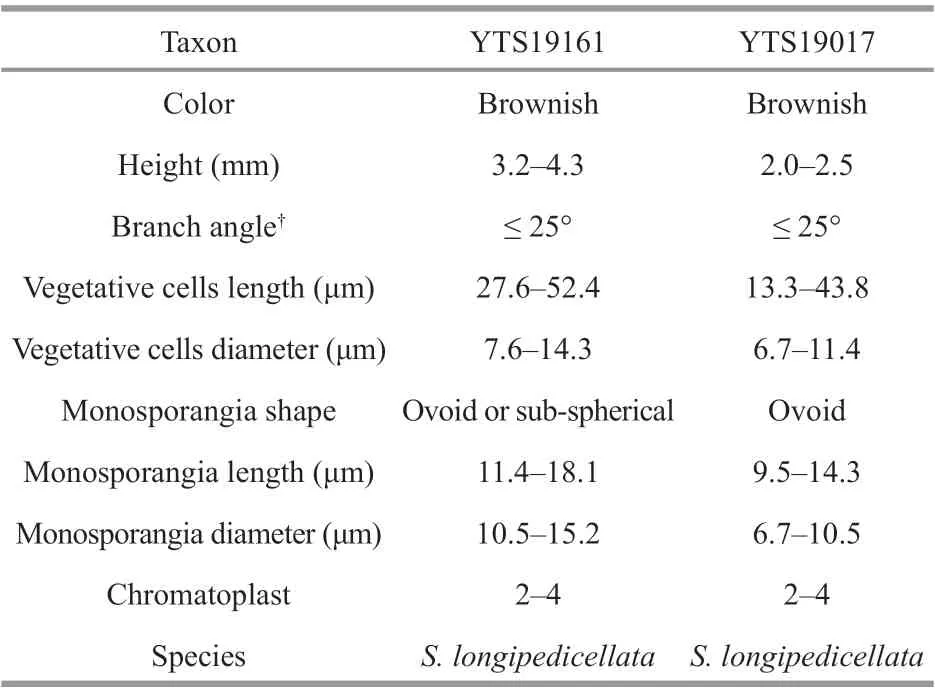
Table 5 Morphological characteristics of “Chantransia”stage specimens in this study
3 RESULT
3.1 Morphological characterization
The morphological characteristics of the four specimens in this study were observed and measured as shown in Fig.1 and Tables 4-5.
The two gametophyte stage specimens (SAS18052 and SAS18523) are in good agreement with the dimension circumscription description ofS.longipedicellata. The SAS18052 specimen displayed a lighter colored thallus with spherical- to barrelshape, adjacent whorls, while the SAS18523 specimen displayed a darker colored thallus with separated and spherical whorls. Furthermore, the carposporophytes of the SAS18052 specimen were within the whorls,while those of the SAS18523 specimen were out of the whorls.
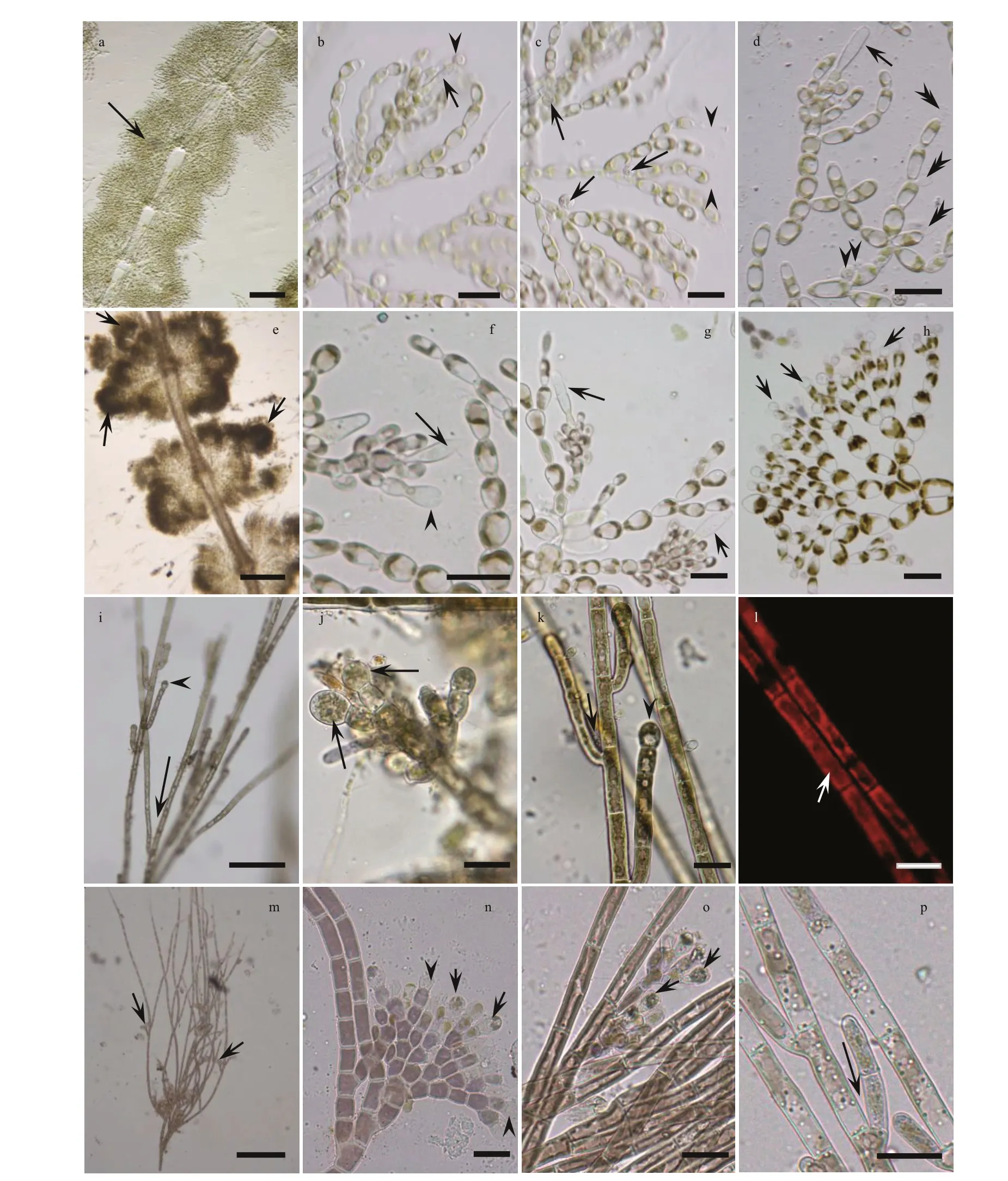
Fig.1 Morphological characters of samples of both gametophyte stage samples and “Chantransia” stage samples investigated in this study
The two “Chantransia” isolates (YTS19161 and YTS19017), were morphologically similar toAudouinellapygmaea(Kützing) Weber Bosse. These two isolates had the following characteristics:microscopic (2.0-4.3 mm) Tuft-shaped with a brownish thallus, composed of creeping and erect f ilaments, narrow branching angles (≤ 25°), cylindrical vegetative cells, and small abundant ovoid or subspherical monosporangia (9.5-18.1 μm in length and 6.7-15.2 μm in diameter).
Then came the Devil once more, and put a different letter in his pocket, in which it was written that they were to put the Queen and her child to death
3.2 Molecular analysis
TherbcL data matrix consisted of 44Sheathiaspecimens (including 20 species), 1 106 characters,of which 452 (40.87%) were variable and 381(34.45%) were parsimony informative. The COI-5P alignment included 33Sheathiasequences (including 17 species) and 571 characters, of which 258 (45.36%)were variable and 223 (39.05%) were parsimony informative. ThepsbA alignment included 21Sheathiasequences (including 9 species) and 834 characters, of which 285 (34.17%) were variable and 253 (30.34%) were parsimony informative sites.Supplementary Tables S3-S5 list the pairwise distance based on the three sequences between all specimens in this study and other species of the order Batrachospermales.
The relationship amongSheathiaspecies and the genusSheathiawith other genera of Batrachospermales based onrbcL, COI-5P, andpsbA alignment, with supporting values calculated from BI and ML methods, are presented in Supplementary Figs.S2-S4. Because we mainly focus on the phylogenetic relationship of genusSheathia, here, only BI trees depicting the relationship amongSheathiaspecies based onrbcL, COI-5P, andpsbA gene sequences are shown with supporting values calculated from the two methods in Figs.2-4. TherbcL and COI-5P data sets include all fourSheathiaspecimens utilized in this study, while thepsbA data set had only three of them (except for YTS19161). Phylogenetic trees of the three markers showed the genusSheathiaforming a distinct, strongly supported clade (Fig.2: 1.00/93.7;Fig.3: 1.00/85.4; Fig.4: 1.00/100).
In therbcL and COI-5P phylogenetic trees, the genusSheathiawas divided into four main clades(Clades I, II, III, and IV; Figs.2-3). However, due to the lack of sequence information in the GenBank database, thepsbA phylogenetic tree was only divided into three clades (Clades I, II, and III; Fig.4). In therbcL phylogenetic tree, Clade I included twelve distinct lineages. From theseS.jiugongshanensisJ.-F.Han, F.-R. Nan & S. L. Xie,S.shimenxiaensisJ.-F.Han, F.-R. Nan & S. L. Xie, andS.qinyuanensisJ.-F.Han, F.-R. Nan & S. L. Xie were only observed in the“Chantransia” sporophyte generations. The remaining nine lineages lacked heterocortication. However, in the COI-5P andpsbA trees, Clade I contained nine and four distinct lineages, respectively. Clade II only containedS.exiguaSalomaki & M. L.Vis, a species that was observed to have heterocortication only on the thickest axis of the thallus near the base of the plant. Clade III was composed of specimens where heterocortication is easy to observe. In therbcL and COI-5P trees, Clade III consists of six closely-related species,S.confusa(Bory) Salomaki & M. L. Vis,S.americanaSalomaki & M. L. Vis,S.boryana(Sirodot) Salomaki & M. L. Vis,S.heterocortica(Sheath & K. M. Cole) Salomaki,S.grandisSalomaki& M. L. Vis, andS.involuta(M. L. Vis & Sheath)Salomaki & M. L. Vis. In contrast, only four of them were included in thepsbA tree. Furthermore, in therbcL and COI-5P trees, Clade IV was consistent with specimens attributed to a new speciesS.californica.The four specimens (both gametophyte stages and“Chantransia” stages included) utilized in this study were within Clade I and clustered with several sequences from China to formed a large clade namedS.longipedicellata, which had high support values except for the Bayesian posterior probabilities (PP)support value in thepsbA tree (Fig.2: 1.00/100; Fig.3:100/99.4; Fig.4: -/96.7).
3.3 Divergence time estimation
Rhodophyta divergence time estimation based on combined sequences (rbcL, COI-5P, andpsbA) matrix(Fig.5) showed that Cyanidiophyceae, Rhoclellophyceae,Compsopogonophyceae, Stylonematophyceae, and Porphyridiophyceae diverged f irst, followed by the divergence of Florideophyceae and Bangiophyceae.Within Florideophyceae, Nemaliophycidae diverged at approximately 662.01 Ma (95% highest posterior density (HPD): 779.83-580.51 Ma) and Batrachospermales and Thoreales orders split occurred at 456.10 Ma (95% HPD: 552.80-367.88 Ma). The divergence time of genusSheathiacan be traced back to 251.49 Ma (95% HPD: 328.07-184.73 Ma).
3.4 Ancestral geographical origin inference
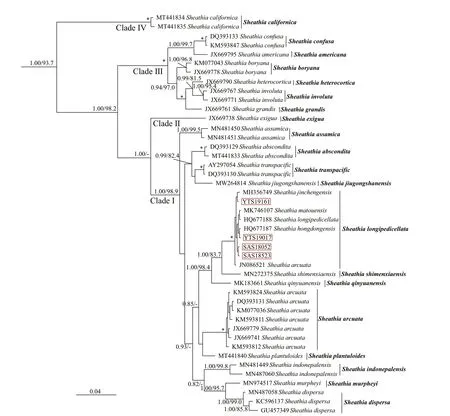
Fig.2 Bayesian inference phylogeny depicting the relationship of among Sheathia species based on rbc L gene sequences
Since we focus primarily on the ancestral geographic origin of the genusSheathia, other unrelated clades were not shown in the results (Fig.6).The ancestral area reconstruction based onrbcL and COI-5P gene data yielded similar results. The biogeographic inference map generated using BBM based on therbcL gene data showed that the most recent common ancestor of the genusSheathiawas in North America, as shown in node 176 (North America,relative probability=0.872 0) (Fig.6a). All specimens in Clade I, including the four samples utilized in this study, showed an Asian distribution at their basal node (node 164, Fig.6a). The Clade I clustering together withS.exiguafrom France has an inferred Asian, European, or North American origin. For species in Clade III with marked heterocortication, a Europe, North America, or Europe + North America distribution at their basal node is shown.
Based on the COI-5P gene, the geographical origin of the genusSheathiawas also traced back to North America, as shown in node 130 (North America,relative probability=0.920 2) (Fig.6b). All samples lacking heterocortication, including the four specimens in this study, were grouped withS.exiguafrom Bulgaria to yield node 127, implying a Europe,North America, or Europe + North America origin.The species that show heterocortication more likely had their origin in North American, as shown in node 106 (North America, relative probability=0.770 4).
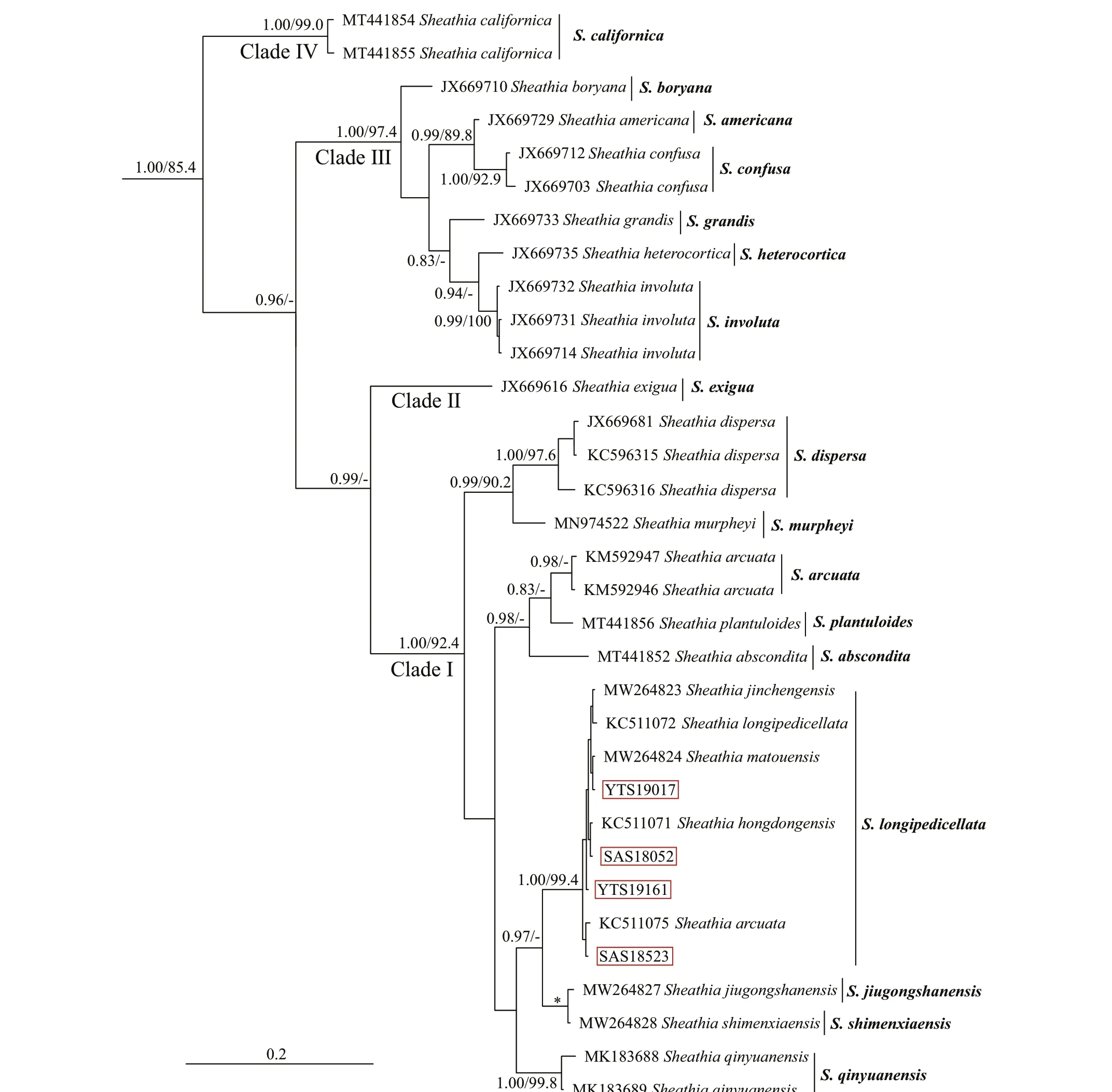
Fig.3 Bayesian inference phylogeny depicting the relationship of among Sheathia species based on COI-5P gene sequences
According to Condie (1982), from the Carboniferous to Jurassic, North America was in the northwest of the Pangaea continent; the Eurasian and the African continents adjoin North America by their west and north, respectively. After its origin in North America, the genusSheathiacould f irst spread to Eurasia and Africa. However,Sheathiataxa in the Oceania spread from African species through India-Antarctica.
4 DISCUSSION
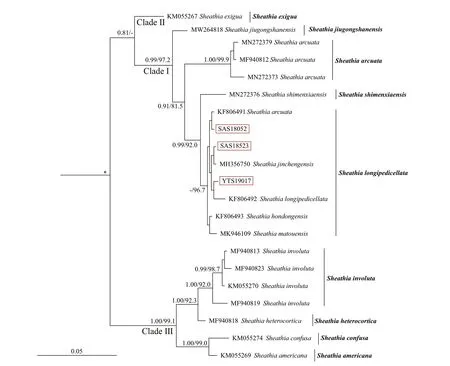
Fig.4 Bayesian inference phylogeny depicting the relationship of among Sheathia species based on psb A gene sequences
Batrachospermumgenus was considered the genus with the most abundant species in the order Batrachospermales (Sheath, 1984; Kumano, 2002).At present, all species of this order are reported in freshwater. The genusBatrachospermumwas classif ied into several sections according to traditional taxonomic assignments and was shown to be paraphyletic since the f irst phylogenetic study of the order Batrachospermales. Recently, sections of this genus have been methodically investigated and raised to separated genera using DNA sequences and morphology (Entwisle et al., 2009; Salomaki et al.,2014; Rossignolo and Necchi, 2016; Necchi et al.,2019a, c; Vis et al., 2020). The genusSheathiawas elevated from the sect.Helminthoideaand was distinguished from other genera by heterocortication(Salomaki et al., 2014). More than twenty species of this genus have been reported worldwide, of whichS.longipedicellatais endemic to China. The morphological observations suggested that most of theSheathiagenus species were diffi cult to distinguish by unique morphological characters. Only specimens ofS.confusaare distinguished from other species by spermatangia on the involucral f ilaments of the carpogonium (Vis and Sheath, 1996; Salomaki et al.,2014). In this study, phylogenetic trees showed that the branch of theSheathiaspecies contained four main clades. Interestingly, species in these four clades contain diff erent cortical cell morphologies:heterocortication can be seen in all species of CladeⅢ, but in the Clade I, Ⅱ, and Ⅳ, heterocortication was either only present on the thickest axis of the thallus near the base or completely absent (Salomaki et al., 2014; Necchi et al., 2019c). Molecular analyses of the three gene regions combined with morphological information conf irmed that SAS18052 and SAS18523 belong toS.longipedicellatawhile the two “pygmaea”isolates YTS19161 and YTS19017 represent the“Chantransia” ofS.longipedicellataand not theAudouinellagenus. Therefore, no correlation was found between the molecular and morphological analysis of the “Chantransia” stage, making this stage an obstacle to species identif ication. Furthermore, the close relationship amongS.longipedicellata,S.hongdongensis(S. L. Xie and J. Feng) J.-F. Han &al.,S.jinchengensisJ.-F. Han & al. andS.matouensisJ.-F. Han, F.-R. Nan, J. Feng, J.-P. Lv, Q. Liu, X.-D.Liu & S. L. Xie has been suggested by previous phylogenetic analyses (Necchi et al., 2019c). Based on the three gene trees, we agree that they are a single species. Therefore, the number ofS.longipedicellatadistribution sites in China expands to two.
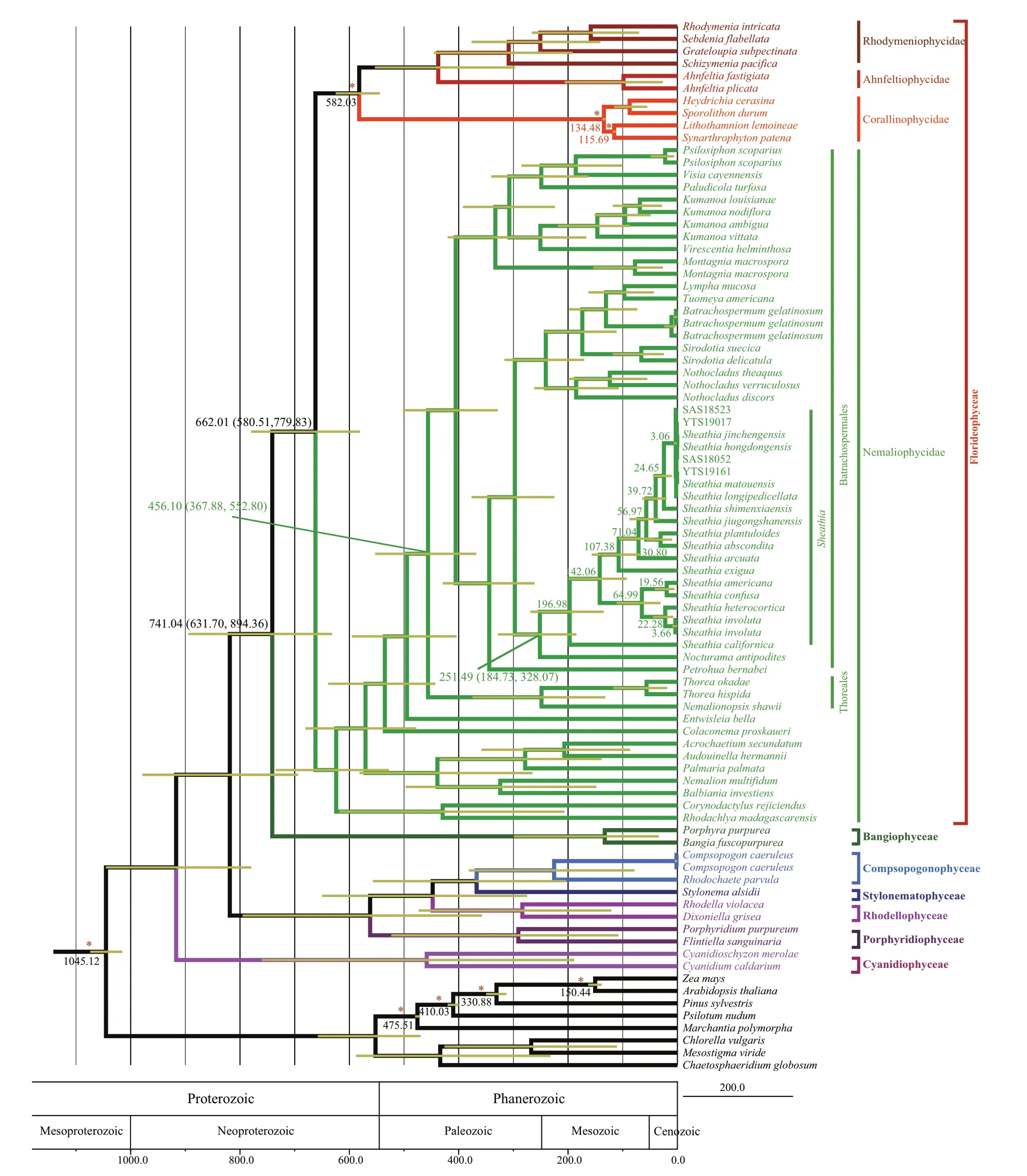
Fig.5 Divergence time estimation phylogram inferred from combined rbc L, COI-5P, and psb A data using BEAST
Although many pieces of research have been carried out onSheathiain recent years, many aspects of the geographical origin and divergence time of this genus remain unresolved. However, several studies on the evolutionary history of Florideophyceae taxa have important reference value for illuminating the origin and evolution of the genusSheathia. Molecularclock estimates of lineage divergence times in the tree of life depend primarily on the oldest recognized fossils reported. However, the age of a fossil only gives a minimum age for the lineage, leading to the uncertainty of the dating of nodes in molecular phylogenetic trees. According to Bengtson et al.(2017),RafatazmiachitrakootensisandRamathalluslobatusbelong to crown-group rhodophytes and were present in the ~1.6 billion years ago (Ga) Vindhyan stromatolitic microbialites. Nevertheless, other studies argued that the morphology ofRafatazmiachitrakootensisandRamathalluslobatusdo not allow for an unequivocal assignment to crown rhodophytes and that they are hundreds of millions of years older than the currently accepted divergence time between rhodophytes and viridiplantae (Betts et al., 2018;Gibson et al., 2018). So far, the fossil red algaBangiomorphapubescensis the oldest known taxonomically resolved eukaryotic. It was f irst described in chert from peritidal carbonate facies of the Hunting Formation on Somerset Island and has long been presumed to arise 1.2 Ga (Butterf ield et al.,1990, 2000).
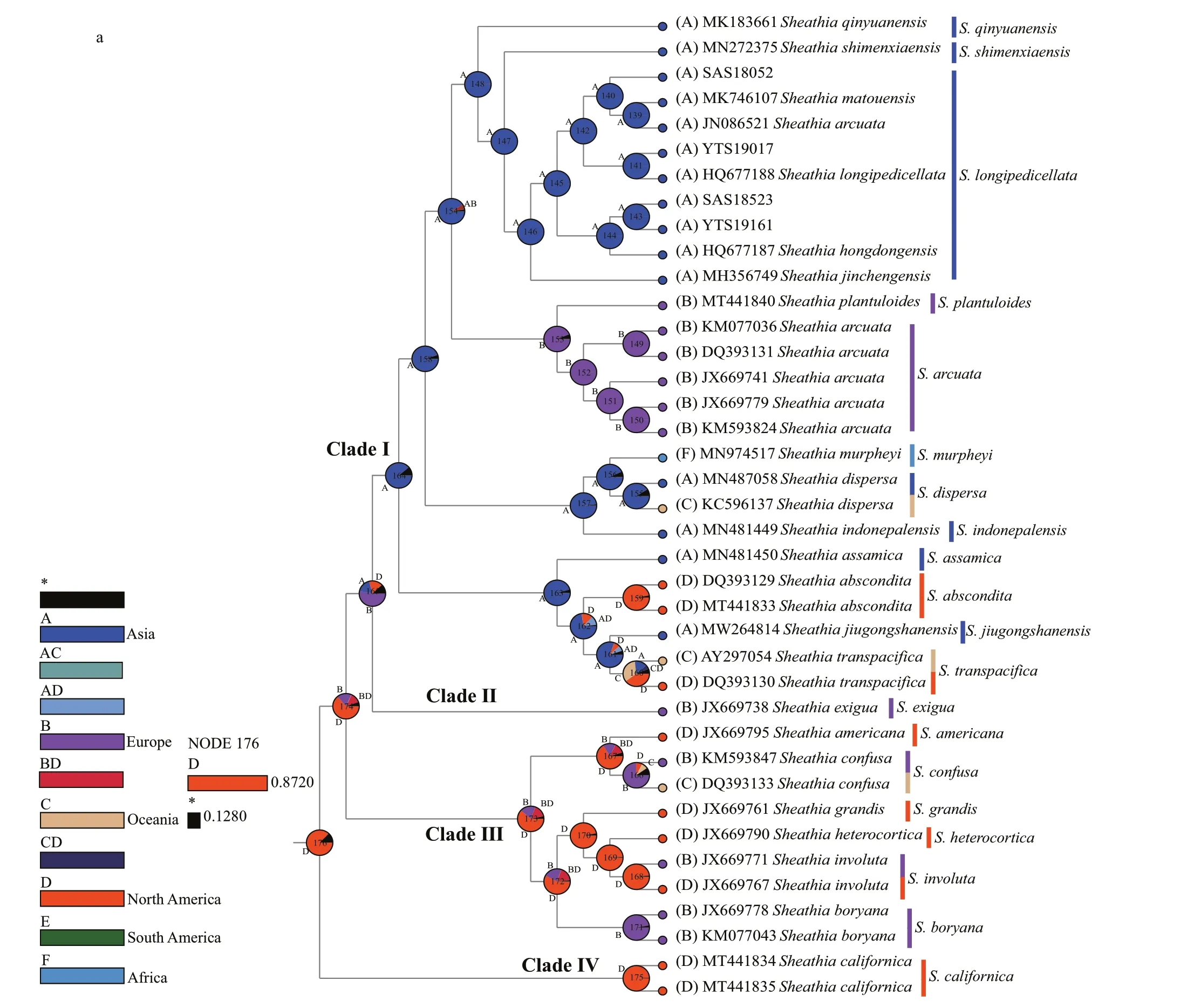
Fig.6 Reconstruction of ancestral distribution area of genus Sheathia based on BBM
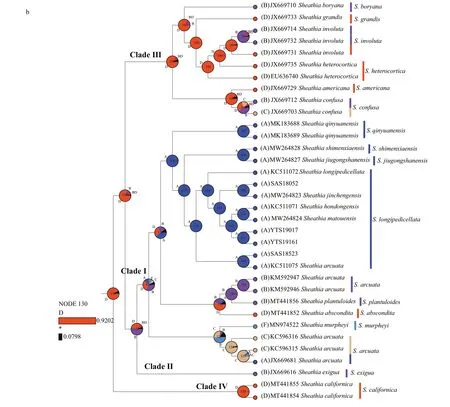
Fig.6 Continued
Nevertheless, several researchers have rejected using the 1.2 GaBangiomorphaas a calibration point for the appearance of bangiophycean or even rhodophyte, because it had seemed too old to make sense (Berney and Pawlowski, 2006; Parfrey et al.,2011; Sharpe et al., 2015). The eukaryote tree of Berney and Pawlowski (2006) showed that whenBangiomorphawas excluded, the origination date of the last ancestor of the eukaryote crown group(LECA) shift from 4 182-1 830 Ma to 1 357-947 Ma.Parfrey et al. (2011) placedBangiomorphaat the base of the crown-group rhodophytes and placed the origin of rhodophytes at 959-625 Ma when Proterozoic fossils were included in the calibration, and 1 285-1 180 Ma when excluded. Similarly, they found that according to whether the Proterozoic fossils were included in the calibration, the divergence time of f lorideophyte and Bangiales was diff erent: when Proterozoic fossils constraints were excluded, they split at ca. 620 Ma (~700-495 Ma), whereas, when those fossils included, their divergence time returns to ca. 765 Ma (915-630 Ma). Sharpe et al. (2015)excludedBangiomorphaas a calibration point and arrived at a rhodophyte/chlorophyte divergence date of 996 Ma (1 186-801 Ma). Yang et al. (2016) and Nan (2017) acceptedBangiomorphaas a rhodophyte;however, they usedBangiomorphaas a calibration point for the early rhodophyte stem group instead of Bangiophyceae and Florideophyceae. Based on a seven-gene concatenated dataset and a Bayesian relaxed-clock model, Yang et al. (2016) suggested that the class Florideophyceae diverged approximately 943 (1 049-817) Ma and Nemaliophycidae diverged approximately 661 (736-597) Ma. Subsequently,divergence time estimation based on chloroplast and mitochondrial genomes indicated that the typical freshwater orders Thoreales and Batrachospermales diverged approximately 484-415 Ma (Nan, 2017).
Bangiomorphapubescenswas also reported in the Angmaat Formation of the Bylot Supergroup on Baffi n Island (Knoll et al., 2013). Based on the Re-Os geochronology analysis, Gibson et al. (2018) revised the f irst appearance ofB.pubescensto 1 047 (+13/-17) Ma and suggested that this date permits more precise estimates of early eukaryotic evolution. Using relaxed molecular clock analysis with the new fossil age as the calibration for rhodophyte and chlorophyte,we suggest that Florideophyceae and Nemaliophycidae originated at Neoproterozoic. Moreover, this study showed that the divergence between orders Batrachospermales and Thoreales occurred during the Early Paleozoic Era to early Late Paleozoic Era (95%HPD: 552.80-367.88 Ma), nearly a million years later than previous studies.
The reconstruction of the geographical area of origin is of great signif icance to understand the evolution and development of red algae. Several studies have been conducted on the biogeography of red algae (Xie, 2001; Feng et al., 2015; Nan et al.,2016). With the help of the distribution of primitive taxa and sister groups, Xie (2001) speculated that the Batrachospermales order might originate in the area connecting the southern edge of Laurentia and the northern edge of Gondwana. Based on geographic information and molecular phylogenetic evidence,Feng et al. (2015) suggested that the genusThoreamost probably originated in Asia. According to Nan et al. (2016), the genusCompsopogonoriginated in North America, which was part of the Laurentia landmass located in the Rodinia supercontinent during the Proterozoic era. Based on the above, we speculate that the genusSheathiashould also originate in the Northern Hemisphere. Based on the comprehensive analysis of the geographic reconstruction and divergence time, we proposed that genusSheathiaoriginated in North America, a portion of the Pangaea supercontinent from the Carboniferous to the Jurassic period (95% HPD: 328.07-184.73 Ma).
In the late Carboniferous, the collision between the Euramerica continent and the Gondwana paleocontinent formed the western half of the Pangaea supercontinent. At the end of Permian, the formation of the Pangaea supercontinent caused most of the oceans formed during the Pannotia supercontinent division to disappear. With the equator as the center,Pangaea extended from the Antarctic to the Arctic.The veritable Pangaea was f inally formed in the Late Triassic (Scotese et al., 1979).
The species of the genusSheathiaare widely distributed in Asia, Europe, North America, Africa,and Oceania, suggesting that the genusSheathiamay have appeared before the Pangaea separated. Divergent time estimates based on a relaxed molecular clock suggested that the genusSheathiaoriginated between the Carboniferous and the Jurassic period. However,according to Condie (1982), by the mid-to-late Jurassic, Africa began to drift away from North America and India-Antarctica. Therefore, the genusSheathiashould appear before this period. Since North America was connected to Eurasia and Africa continent in Pangaea (Scotese et al., 1979),Sheathiataxa could directly expand to these two continents after their origin in North America. Nevertheless, Oceania does not directly border North America in Pangaea (Scotese et al., 1979). Hence we inferred that theSheathiaspecies of Oceania were spread from Africa.
5 CONCLUSION
Using morphological characteristics and genetic data (rbcL, COI-5P, andpsbA), we analyzedSheathiagenus samples from the Shanxi (gametophyte stages)and Henan (“Chantransia” stages) provinces.Morphological data show that the “Chantransia” stage samples (YTS19161 and YTS19017) were similar toA.pygmaea, while the gametophyte stage samples(SAS18052 and SAS18523) were highly consistent with the description ofS.longipedicellata.Conforming to the morphological identif ication results, all three molecular markers supported SAS18052 and SAS18523 asS.longipedicellata.Nevertheless, phylogenetic trees indicated that YTS19161 and YTS19017 are the “Chantransia” ofS.longipedicellatarather than the genusAudouinella.Moreover, the evolutionary history ofSheathiashowed that it had originated about 328.07-184.73 Ma in the portion of Pangea that was to become North America, then spread to other localities.
6 DATA AVAILABILITY STATEMENT
The data that support the f indings of this study are available on request from the corresponding author.
7 ACKNOWLEDGMENT
Dr. N. R. C. Eva (University of Puerto Rico, USA)is acknowledged for the English editing.
 Journal of Oceanology and Limnology2022年2期
Journal of Oceanology and Limnology2022年2期
- Journal of Oceanology and Limnology的其它文章
- Identif ication of Antarctic minke and killer whales with passive acoustic monitoring in Prydz Bay, Antarctica*
- Eff ects of dissolved oxygen and nutrients from the Kuroshio on hypoxia off the Changjiang River estuary*
- Methane in the Yellow Sea and East China Sea: dynamics,distribution, and production*
- Longitudinal genetic analysis of growth-related traits in red swamp crayf ish Procambarus clarkii (Girard)*
- Early life migration and population discrimination of the small yellow croaker Larimichthys polyactis from the Yellow Sea: inferences from otolith Sr/Ca ratios*
- A new oil spill detection algorithm based on Dempster-Shafer evidence theory*