
Contrasting vertical distribution between prokaryotes and fungi in diff erent water masses on the Ninety-East Ridge,Southern Indian Ocean*
2022-04-07 09:09ShujunLIZhisongCUIMutaiBAOXiaoLUANFeiTENGShujiangLITengfeiXULiZHENG
Shujun LI , Zhisong CUI , , Mutai BAO , Xiao LUAN , Fei TENG , Shujiang LI ,Tengfei XU , Li ZHENG
1 Frontiers Science Center for Deep Ocean Multispheres and Earth System, Key Laboratory of Marine Chemistry Theory and Technology, Ministry of Education, and College of Chemistry and Chemical Engineering, Ocean University of China, Qingdao 266100, China
2 Marine Bioresources and Environment Research Center, First Institute of Oceanography, Ministry of Natural Resources of China, Qingdao 266061, China
3 Laboratory for Marine Ecology and Environmental Science, Pilot National Laboratory for Marine Science and Technology(Qingdao), Qingdao 266237, China
4 State Key Laboratory of Environmental Aquatic Chemistry, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China
Abstract Although the microbial diversity of the Indian Ocean has been extensively investigated, little is known about the community composition of microbes in the Southern Indian Ocean. In the present study, we divided 60 water column samples on the Ninety-East Ridge (NER) into f ive water masses according to the temperature-salinity curves. We presented, for the f irst time, a full description of the microbial biodiversity on NER through high-throughput amplicon sequencing approach, including bacteria, archaea, and fungi. We found that bacteria exhibited higher richness and diversity than archaea and fungi across the water masses on NER. More importantly, each water mass on NER featured distinct prokaryotic microbial communities,as indicated by the results of non-metric multidimensional scaling. In contrast, fungi were eurybathic across the water masses. Redundancy analysis results demonstrated that environmental factors might play a pivotal role in the formation and stability of prokaryotic communities in each water mass, especially that of archaea.In addition, indicator species might be used as f ingerprints to identify corresponding water masses on NER.These results provide new insights into the vertical distribution, structure, and diversity of microorganisms on NER from the perspective of water mass.
Keyword: Ninety-East Ridge (NER); temperature-salinity curve; microbial vertical distribution; water mass; microbial diversity
1 INTRODUCTION
Microbes play a pivotal and fundamental role in the marine food web and global nutrient cycles(Arrigo, 2005; Fuhrman et al., 2015), participating in almost all oceanic biogeochemical processes. It is necessary to study the biodiversity of marine microbes before understanding their ecological role and function. To date, microbial diversity of the Indian Ocean has been extensively investigated (Sinha et al.,2019; Wang et al., 2021). However, little is known about the community composition of microbes in the Southern Indian Ocean. For instance, bacterial distribution in the deep-sea sediments of the Central Indian Ridge and the equator region of the Indian Ocean have been investigated (Hoek et al., 2003;Khandeparker et al., 2014). Qian et al. (2018)characterized the composition of bacterial and archaeal communities from rare earth elements-rich sediments at a depth of approximately 4 800 m in the Indian Ocean by Illumina high-throughput sequencing targeting 16S rRNA genes.
The mid-ocean ridges are unique ecosystems in terms of the structure of the benthic and pelagic communities they harbor, as well as the relatively high biomass of some ridges (Ingole and Koslow,2005; Ramirez-Llodra et al., 2010). These ridges represent the current research hotspots (Ramirez-Llodra et al., 2010). Ninety-East Ridge (NER) is an earthquake-free ridge in the south-north direction along the line of 90°E in the Indian Ocean (Krishna et al., 1999). It is part of the mid-ocean ridge. In addition,NER is one of the most complicated regions exhibiting unique tectonic, topographic, bathymetric, and hydrographic features (Gao et al., 2021). These factors inf luence the distribution and ultimately the structuring of bacterial communities in water (Qian et al., 2018). However, it remains unclear how microbial communities respond diff erently to the changes in environmental conditions along the vertical gradient of various depths. Therefore, the study of the microbial community composition and biodiversity of the rarely-explored water column on NER in Indian Ocean can provide new insights into the structure and function of this unique bathypelagic ecosystem.
A water mass is a large body of water with relatively uniform physical, chemical, and biological characteristics and generally the same changing trends that exhibits signif icant diff erences from the surrounding seawater (Sverdrup et al., 1942; Yoshida et al., 1971). A water mass def ines the diff usion boundary of the matter in it. Previous investigators used water masses to study the vertical microbial diversity of pelagic ecosystems. For instance, Lovejoy et al. (2006) analyzed the microbial eukaryote diversity of f ive diff erent water masses in the Arctic Ocean using 18S rRNA gene clone library. In addition,another previous study characterized the taxonomic and functional community diversity of diff erent water masses in the Southwestern Atlantic Ocean (SAO)(Junior et al., 2015). Compared with the microbial community diversity of the global ocean, the SAO harbored a signif icant portion of endemic gene diversity (Junior et al., 2015).
Improving our knowledge of the biodiversity of all microbial species is of great importance because ubiquitous and extensive interplay exists among diff erent types of microbes so that they function as a whole to participate in important biogeochemical processes. However, almost all studies to date have only investigated a specif ic category of microorganisms rather than the whole microbial community, including bacteria, archaea, and fungi. For instance, Li et al.(2019a) assessed the depth-related distribution pattern of fungal communities along the water columns from the epi- to abyss-opelagic zones of the Western Pacif ic Ocean using internal transcribed spacer 2 (ITS2)metabarcoding. In addition, a previous study presented the f irst “snapshot” on biodiversity and spatial distribution of the bacteria in water columns in the Eastern Indian Ocean. This study suggested a more pronounced stratif ied distribution pattern with a detailed description of the bacterial community structure by pyrosequencing (Wang et al., 2016).
In view of the internal homogeneity of the water mass, we proposed the hypothesis that distinct microbial assemblage (including bacteria, archaea,and fungi) existed in diff erent water masses on NER,and the microbial community structures were mainly controlled by the corresponding environmental factors of the water masses. In the present study, we f irst divided the water column samples from NER into f ive water masses according to the temperaturesalinity curves (T-Scurves). Then, we used highthroughput amplicon sequencing approach and bioinformatics to present a full and comparative description of the microbial community structure and biodiversity on NER, including bacteria, archaea, and fungi.
2 MATERIAL AND METHOD
2.1 Sampling and demarcation of water masses by T- S curves
Seawater was sampled with a CTD rosette (Seabird 911, SeaBird Electronics, Bellevue, Washington,USA) at 12 stations on NER (Fig.1a) from Dec. 2018 to Jan. 2019. Five samples were taken at depths ranging from 5 m to 5 000 m at each station. After sampling, the f iltration has been done with a f ilter bowl (PALL, EastHills, NY, USA) and a diaphragm pump (Jinteng, Tianjin, China) on board. For epipelagic waters (depth <1 000 m), 2 L of seawater was f iltered through a 0.22-μm pore-size membrane(Whatman, 47-mm diameter) at -0.08 MPa to collect microbial biomass. For bathypelagic waters (depth>1 000 m), 10 L of water was pretreated by the same method. Then, the membranes with biomass were kept at -80 °C on board prior to DNA extraction.
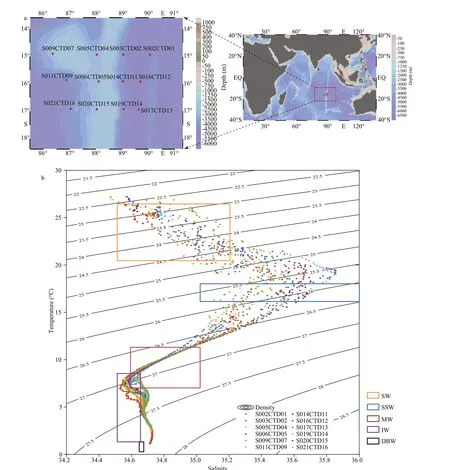
Fig.1 Investigation sites and diff erent water masses on NER
T-Scurves were generated from the physical oceanographic data of these 12 stations using Matlab(version R2010a) immediately after sampling. The prof ile of theT-Scurves indicated the presence of f ive diff erent water masses (Fig.1b; Table 1). Accordingly,these 60 water samples were sorted into 5 water masses in order to analyze the vertical distribution of the microbial community (Hanawa and Talley, 2001;Yao et al., 2017).
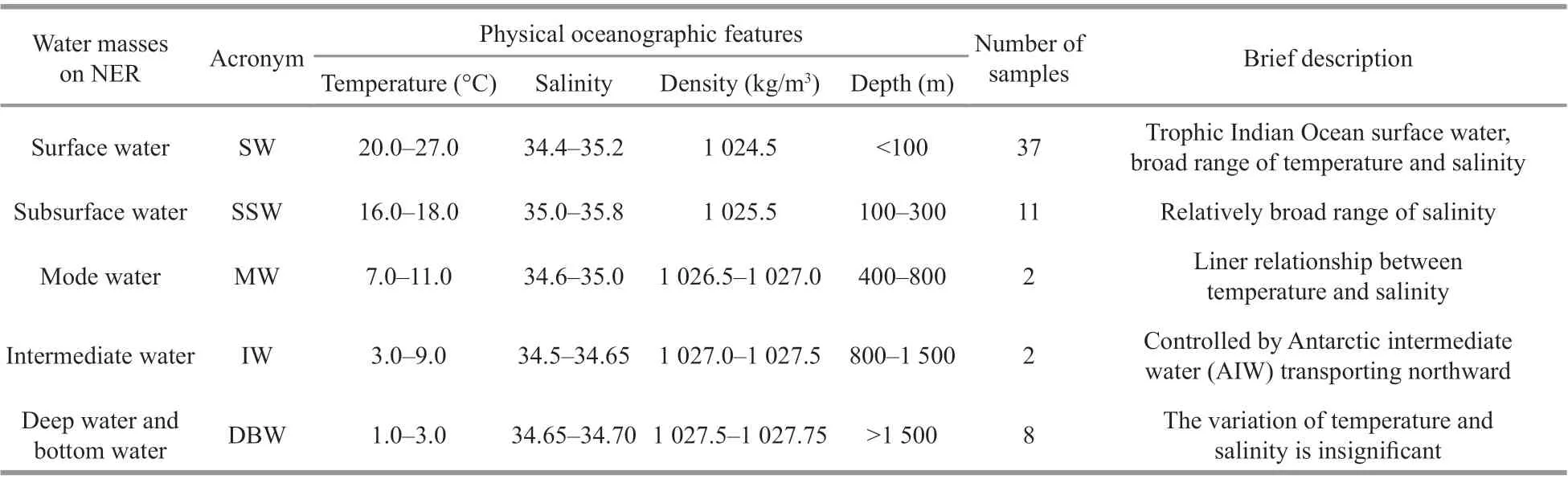
Table 1 Five water masses on NER and their physical oceanographic features
2.2 Physical and chemical parameters
At each sampling station, a CTD prof iler was deployed to record temperature, depth, salinity, pH,and dissolved oxygen prof iles. Samples for the analysis of dissolved nutrients (nitrate, phosphate,and silicate) were f iltered onto Whatman GF/F f iberglass f ilters, and stored at -20 °C. The chemical parameters of seawaters with depth less than or equal to 500 m were analyzed as described by Park (1969),while those of seawaters with depth greater than 500 m were downloaded from GLODAPv2 data (Key et al., 2015; Lauvset et al., 2016; Olsen et al., 2016).
2.3 Total genomic DNA extraction
Total genomic DNA was extracted from the membrane samples described in Section 2.1 with Fast DNA SPIN extraction kits (MP Biomedicals, Santa Ana, CA, USA). Afterwards, DNA concentration was measured by NanoDrop ND-1000 spectrophotometer(Thermo Fisher Scientific, Waltham, MA, USA). The DNA molecular size was estimated by 0.8% (w/v)agarose gel electrophoresis. Then, the extracted DNA samples were stored at -80 °C.
2.4 High-throughput amplicon sequencing and data analysis
The V3-V4 region of the 16S rRNA gene was amplif ied from the bacterial DNA by PCR using the forward primer 5′-ACTCCTACGGGAGGCAGCA-3′and the reverse primer 5′-GGACTACHVGGGTWTCTAAT-3′ (Claesson et al., 2009). The V4-V5 region of the 16S rRNA gene was amplif ied from the archaeal DNA by PCR using the forward primer 5′-TGYCAGCCGCCGCGGTAA-3′ and the reverse primer 5′-YCCGGCGTTGAVTCCAATT-3′ (Pires et al., 2012). The ITS1 region was amplif ied from the fungal DNA by PCR using the forward primer 5′-GGAAGTAAAAGTCGTAACAAGG-3′ and the reverse primer 5′-GCTGCGTTCTTCATCGATGC-3′(White et al., 1990). Sample-specif ic 7-bp barcodes were incorporated into the primers for multiplex sequencing. The PCR components contained 5 μL of buff er (5×), 0.25 μL of Fast Pfu DNA Polymerase(5 U/μL), 2 μL (2.5 mmol/L) of dNTPs, 1 μL(10 μmol/L) of each forward and reverse primer, 1 μL of DNA Template, and 14.75 μL of ddH2O. Thermal cycling consisted of initial denaturation at 98 °C for 5 min, followed by 25 cycles consisting of denaturation at 98 °C for 30 s, annealing at 53 °C for 30 s, and extension at 72 °C for 45 s, with a f inal extension of 5 min at 72 °C. PCR amplicons were purified with Vazyme VAHTSTM DNA Clean Beads (Vazyme,Nanjing, China), and quantified using the Quant-iT PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad,CA, USA). After the individual quantif ication step,amplicons were pooled in equal amounts. Then, the sequencing library was prepared using TruSeq Nano DNA LT Library Prep Kit from Illumina (San Diego,California, USA). Finally, pair-end 2× 250 bp sequencing was performed using the Illumina NovaSeq platform with NovaSeq 6000 SP Reagent Kit (500 cycles) at Shanghai Personal Biotechnology Co., Ltd. (Shanghai, China).
QIIME 2 (Bokulich et al., 2018; Bolyen et al.,2019) was used to do downstream analysis of raw sequence data generated by PE250. Brief ly, raw sequence data were demultiplexed using the demux plugin followed by primers cutting with the cutadaptplugin (Martin, 2011). Sequences were then quality f iltered, denoised, merged, and chimera removed using the DADA2 plugin (Callahan et al., 2016)(Supplementary Tables S1-S3). Non-singleton amplicon sequence variants (ASVs) were aligned with maff t (Kazutaka et al., 2002) and used to construct a phylogeny with fasttree2 (Price et al.,2009). Bacterial samples were raref ied to 26 600 sequences per sample. Archaeal samples were raref ied to 34 800 sequences per sample. Fungal samples were raref ied to 44 920 sequences per sample. Alphadiversity metrics, including Chao1 (Chao, 1984),Observed species, Shannon (Shannon, 1948),Simpson (Simpson, 1949), Pielou’s evenness (Pielou,1966), and Good’s coverage (Good, 1953), and beta diversity metrics (Jaccard distance) were estimated using the diversity plugin. Taxonomy was assigned to ASVs using the classify-sklearn naï ve Bayes taxonomy classif ier in feature-classif ier plugin(Bokulich et al., 2018) against the SILVA Release 132(bacteria and archaea) /UNITE Release 8.0 (fungi)Database (Kõljalg et al., 2013).
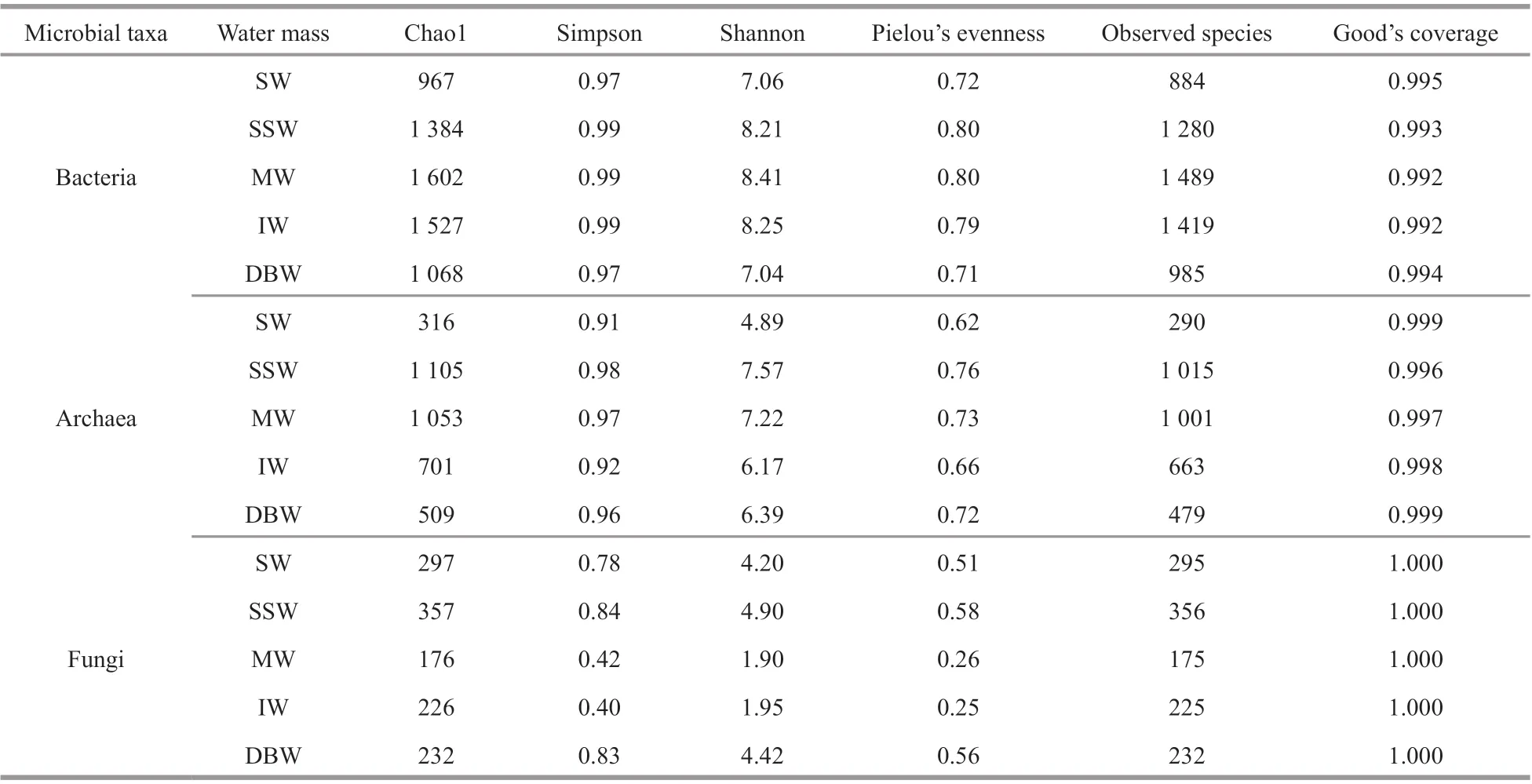
Table 2 Alpha diversity indices of microbial communities in diff erent water masses on NER
2.5 Statistic analysis
Sequence data analyses were mainly performed using QIIME2 and R packages (v3.2.0). We used nonmetric multidimensional scaling (NMDS) to describe the distribution characteristics of samples with Jaccard distance. We also used an analysis of similarities (ANOSIM) to compare microbial community structures among diff erent water masses.Redundancy analysis (RDA) was used to test the relationships between environmental parameters and microbial communities to extract principal environmental factors inf luencing the microbial communities. ThePvalue of the RDA was obtained by Monte Carlo substitution test. A linear discriminant analysis eff ect size (LEfSe) (Segata et al., 2011) was carried out to determine indicator species in the microbial community.
3 RESULT AND DISCUSSION
3.1 Alpha diversity of microbial communities in diff erent water masses on NER
The alpha diversity of microbial communities on NER was compared among water masses (Table 2).Good’s coverage of microbial communities in each water mass was almost 1.000, indicating that the sequencing data was suffi cient to ref lect the microbial community diversity in the f ive water masses on NER.
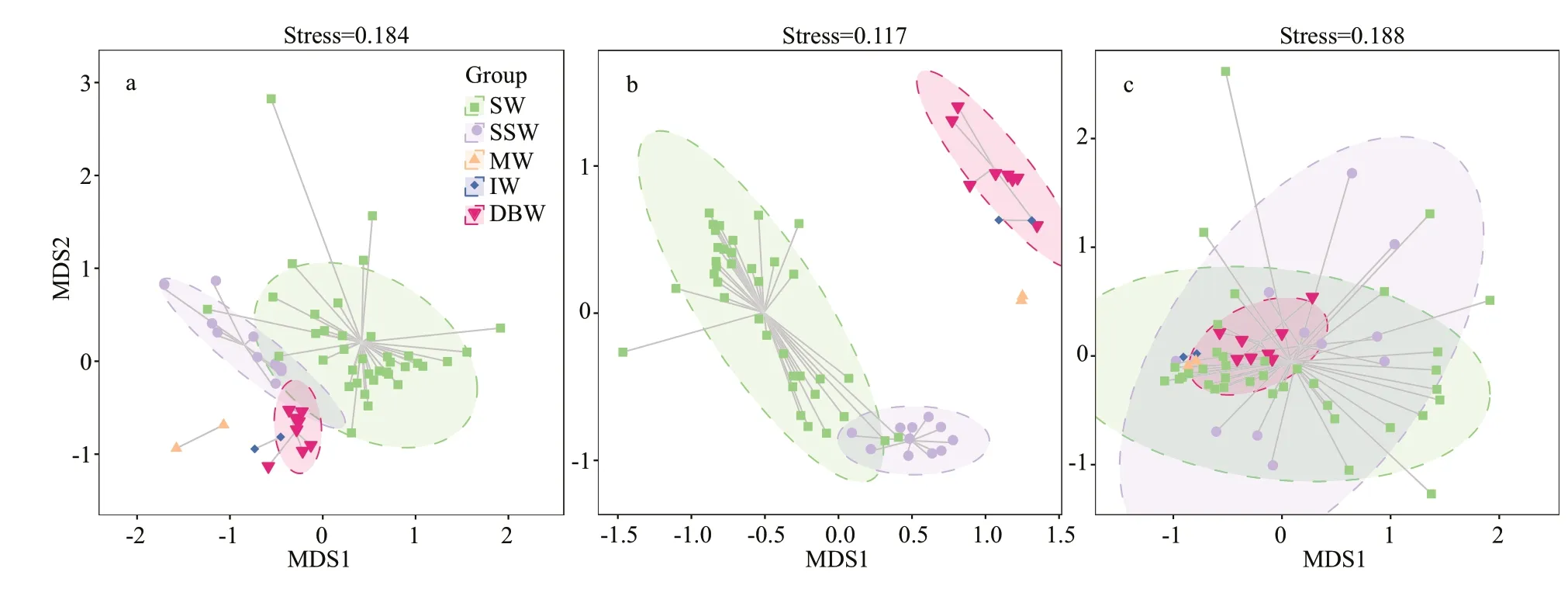
Fig.2 The dissimilarity of microbial community structure among the f ive water masses on NER by NMDS analysis
From the perspective of species, bacteria exhibited the highest richness (Chao1: 967-1 602; observed species: 884-1 489), followed by archaea (Chao1:316-1 105; observed species: 290-1 015) (Table 2).Fungi exhibited the lowest richness (Chao1: 176-357;observed species: 175-356). Similarly, bacteria exhibited the highest diversity (Shannon, 7.04-8.41;Simpson, 0.97-0.99), followed by archaea (Shannon,4.89-7.57; Simpson, 0.91-0.98). Fungi exhibited the lowest diversity (Shannon, 1.90-4.90; Simpson,0.40-0.84). Bacteria also exhibited the highest evenness (0.71-0.80), followed by archaea (0.62-0.76). Fungi exhibited the lowest evenness (0.25-0.58). Furthermore, a dissimilarity analysis based on one-way analysis of variance (ANOVA) showed that microbial alpha diversity in the water column on NER was signif icantly diff erent among bacteria, archaea,and fungi (Supplementary Table S4) (P<0.01). In brief, the alpha diversity of bacteria in the water column on NER was highest, followed by that of archaea. In contrast, fungi exhibited the lowest alpha diversity.
3.2 Beta diversity of microbial communities in diff erent water masses on NER
NMDS analysis was conducted to demonstrate the diff erence among the microbial community structures in each water mass (Fig.2). The result of NMDS analysis was relatively reliable for bacteria, archaea,and fungi (stress value <0.2) (Legendre and Legendre,1998).
The distance between two points on the plot indicated the discrepancy of the microbial community structures between the corresponding two water samples. With respect to bacterial and archaeal community structure, the samples derived from the same water mass formed a distinct and separate cluster on the plot. The discrepancy within the samples from the same water mass was lower than those of the samples derived from diff erent water masses. In contrast, the fungal community structure within the samples of the same water mass could exhibit even higher discrepancy than that of the water samples derived from diff erent water masses. In addition, we performed an analysis of similarities(ANOSIM) of microbial community structure among the f ive water masses on NER (Supplementary Table S5). Both the bacterial and archaeal community structures exhibited signif icant diff erences among diff erent water masses on NER (P<0.01). In contrast,the fungal community structures exhibited no signif icant diff erences among diff erent water masses on NER (P>0.05).
In the open oceans, dispersal of bacteria and archaea is relatively passive (Winter et al., 2013),suggesting that the movement of prokaryotes might be constrained by the interface of water mass.Diff erent water masses exhibit distinct physical oceanographic and chemical features, which are optimum for microbes with the specif ic traits to grow.Therefore, distinct prokaryotic community structures were stratif ied in corresponding water masses on NER, especially that of archaea (Fig.2; Supplementary Table S5). In another previous study, Celussi et al.(2010) analyzed bacterial community structure of f ive Ross Sea water masses using denaturing gradient gel electrophoresis. The results showed that diff erent water masses on Ross Sea also harbored diverse bacterial communities.
In contrast, fungal species were eurybathic in diff erent water masses on NER, which was consistent with previous f indings on the distribution pattern of fungal species in the deep sea (Li et al., 2019a).Moreover, Wang et al. (2019) assessed the diversity and distribution of fungal communities from the Mariana Trench with a depth range of 1 000-4 000 m using Illumina Hiseq sequencing. The results indicated that there were many fungal taxa resources in the seawater of the Mariana Trench, and most of the same fungal species were found at various depths.
In short, the changing environmental factors across water masses might play a key role in the formation and stabilization of prokaryotic microbial communities, but exerted limited impact on fungal community distribution.
3.3 Microbial community structure in diff erent water masses on NER
Each water mass on NER is distinct with respect to the physical oceanographic features (Table 1), which favor or inhibit the growth of specif ic microbes living in the water masses (Guo et al., 2018). Therefore,diff erent water masses usually harbor signif icantly diff erent microbial communities. We looked into the species composition of the microbial communities in the f ive water masses on NER by sequencing and alignment of 16S rRNA and ITS1 genes (Fig.3).
From the perspective of bacterial species composition at the level of phylum (Fig.3a),Proteobacteria was predominant among the bacterial ASVs (>59.0%), which was consistent with previous f indings on oceanic bacterial diversity (Medina-Silva et al., 2018). Actinobacteria, Bacteroidetes,Chlorof lexi, Deinococcus-Thermus, Acidobacteria,and Marinimicrobia (SAR406 clade) were backbone members across the f ive water masses on NER, though the relative abundance of them varied from water mass to water mass. More specif ically, Cyanobacteria are capable of performing photosynthesis and nitrogen f ixation in the presence of light and oxygen (Herrero et al., 2001). Therefore, Cyanobacteria were the more abundant member in SW (9.7%) with available sunlight. The salinity range in SSW is relatively broader. Chlorof lexi and Acidobacteria exhibit adaptability to wide range of changing salinity. Hence,these two phyla prevailed in SSW (Zhao et al., 2020).We found abundant Chlorof lexi and Marinimicrobia(SAR406 clade) in MW and IW, because these two phyla mainly exist in the transition phase from the euphotic to the aphotic layers (Korlević et al., 2015).In addition, members of Actinobacteria thrive in DBW,because they adapt more easily to hostile environmental conditions in the deep ocean (Sayed et al., 2020). Each water mass harbors several dominant bacterial phyla that adapt best to the corresponding environmental conditions of the water mass.
Regarding archaea, Euryarchaeota and Thaumarchaeota were the two predominant phyla in each water mass (Fig.3b). They have been detected in all the oceans of the world, and are ubiquitous from surface waters to the deep sea (Pereira et al., 2019).We found that Euryarchaeota was most predominant in SW, while its relative abundance dropped dramatically with increasing depth (from 80.6% in SW to 32.1% in the other four water masses). In contrast, the abundance of Thaumarchaeota was relatively lower in SW than in the rest of the water masses, but it surpassed Euryarchaeota as the most dominant archaeal phylum with increasing depth(from 18.5% in SW to 67.8% in the other four water masses). In addition, other phyla of low relative abundance (<0.1%) were also present in the water masses (Supplementary Table S6). In summary, our study reinforced previous f indings by high-throughput amplicon sequencing that Euryarchaeota and Thaumarchaeota represented the majority of the sequences in the archaeal communities from extensive regions of the world’s oceans, including the Pacif ic Ocean, the South China Sea, and Jeju Island (Choi et al., 2016; Xia et al., 2017; Li et al., 2019b).
Regarding fungi, Ascomycota (41.1%-95.1%) was the single dominant phylum across the f ive water masses, followed by Basidiomycota (0.6%-5.4%)(Fig.3c). Moreover, there were also several phyla exhibiting low relative abundance (<0.07%) in the water masses (Supplementary Table S7). This prof ile at the phylum level was identical to that of the Central Indian Ocean Basin, which was described in the f irst report on culture-independent diversity of fungi from the Indian Ocean (Singh et al., 2011). Our results reinforced previous f indings that Ascomycota is the largest and most widespread phylum of fungi in deepsea environments (Nagano and Nagahama, 2012;Xu et al., 2014; Li et al., 2019a).
From the perspective of bacterial species composition at the level of genus (Fig.3d),CandidatusActinomarinawas the dominant genus in SW.Sva0996 marine group, also an actinobacterial taxa,was predominant in MW. Actinobacteria can assimilate phytoplankton-derived dissolved protein and are involved in the cycling of dissolved organic nitrogen in the ocean (Orsi et al., 2016). Three bacterial genera (Thermus,Halomonas, andLeeuwenhoekiella) were predominant in IW. Strains ofLeeuwenhoekiellaspp. are pressure-resistant, and have been isolated and characterized from the deep sea environment many times (Liu et al., 2016).LimnobacterandMethyloversatiliswere the dominant genera in the DBW.Limnobacteris the dominant species of DBW owing to its psychrotolerant features(Maity et al., 2012).
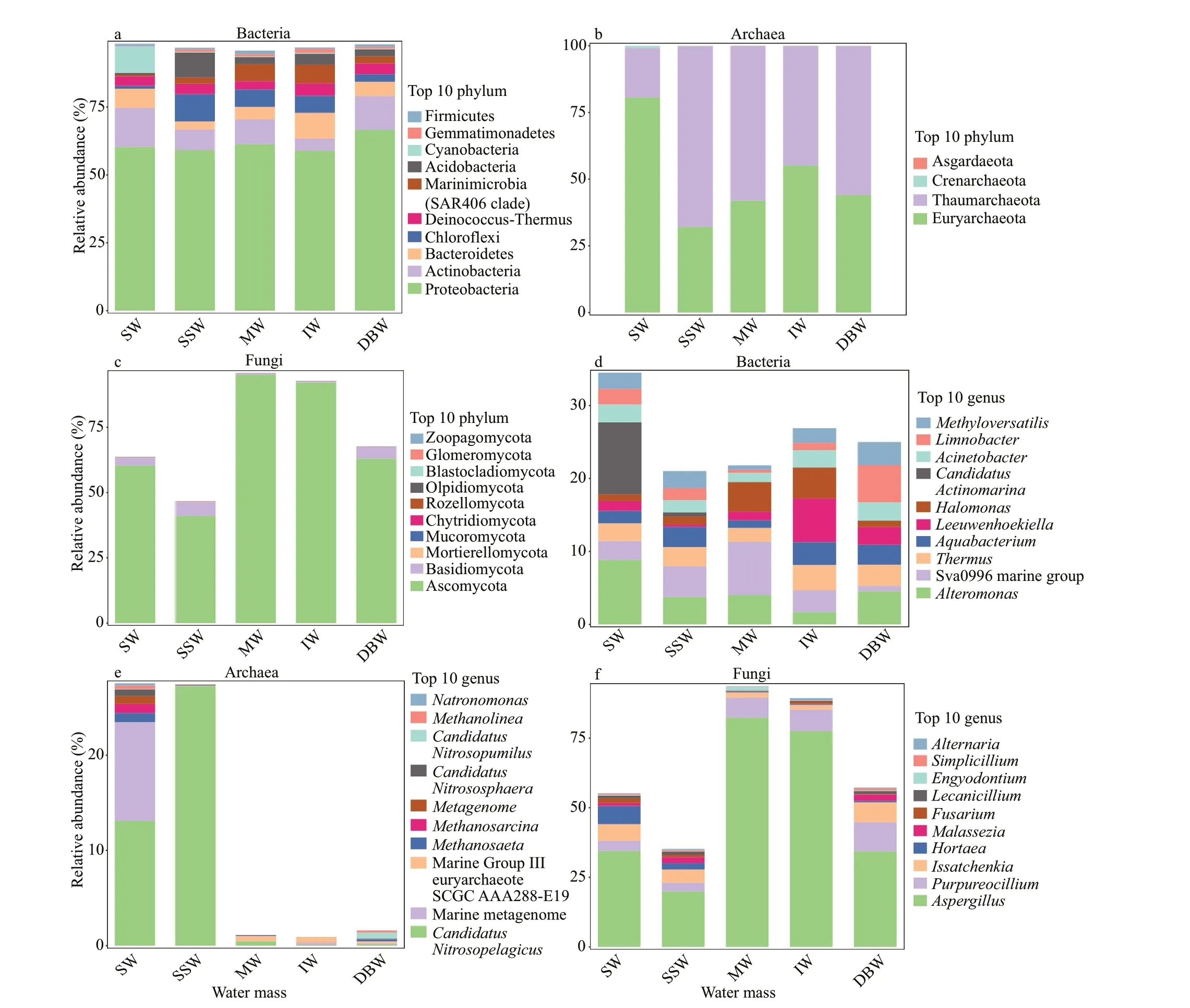
Fig.3 Relative abundance of the top 10 dominant ASVs in the f ive water masses at the level of phylum and genus
It was readily apparent that the relative abundance of archaeal genera in three water masses (MW, IW,and DBW) was surprisingly low (Fig.3e). This prof ile was generated for two reasons: 1) most archaeal ASVs in MW, IW, and DBW could not be identif ied at the genus level, and 2) only the identif ied archaeal genera were represented in the bar graph. Therefore,most unidentif ied members were not represented even though they might be abundant in the corresponding water mass.CandidatusNitrosopelagicuswas the dominant archaeal member in SW. Similarly,Ca.Nitrosopelagicuswas the single dominant archaeal member in SSW. In addition, Marine Group III euryarchaeote SCGC AAA288-E19 was the dominant archaeal member in MW and IW.Ca.Nitrosopumiluswas dominant in the DBW.Ca.NitrosopelagicusandCa.Nitrosopumilusare both ammonia-oxidizing archaea (Park et al., 2012; Carini et al., 2018). The identif ied lineages of this phylum are known to be ammonia oxidizers that might play a signif icant role in natural biogeochemical cycles such as nitrogen cycling (Pester et al., 2011; Hatzenpichler, 2012).
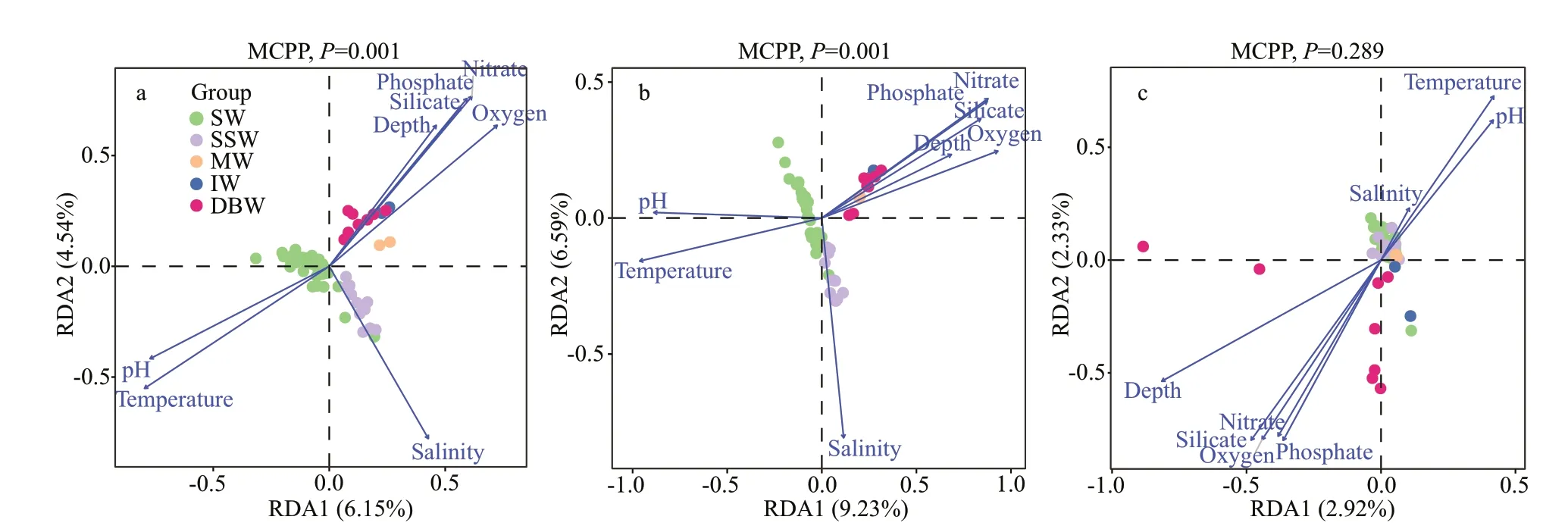
Fig.4 Redundancy analysis of the eff ect of environmental factors on microbial community structure in diff erent water masses on NER
The top 10 most abundant fungal genera made up 66.15% of the fungal communities in the water column on NER (Fig.3f). This proportion was much higher than that of bacteria and archaea, indicating a less complex fungal community structure. The relative abundance ofAspergilluswas the highest across the f ive water masses, especially in MW and IW (>78%). The next predominant genera werePurpureocilliumandIssatchenkia.Aspergilluswas among the most common fungal genera in deep-sea ecosystems (Nagano and Nagahama, 2012). The class Archaeorhizomycetes, which includes most characterizedAspergillusspecies, exhibits its broad distribution across diverse ecosystems and its high species diversity within sites (Rosling et al., 2013).Rosling et al. (2011) formally described it as comprising hundreds of cryptically reproducing f ilamentous species that do not form recognizable mycorrhizal structures and have a saprotrophic lifestyle.
3.4 Environmental factors aff ecting the structure of microbial communities in the diff erent water masses on NER
The physical and chemical parameters of 60 seawater samples derived from NER were summarized in Table 1. Then, RDA was performed between the structure of microbial communities in water masses on NER and the corresponding environmental parameters to explore their correlations (Fig.4). It can be easily seen that environmental factors exerted signif icant impacts on the structure of bacterial and archaeal communities in each water mass (P=0.001)(Fig.4a-b). On the contrary, the same environmental factors exerted insignif icant impacts on the structure of fungal communities in each water mass (P>0.05)(Fig.4c). In order to further determine the variables which signif icantly inf luence microbial community structure, a forward selection procedure (Uraibi et al.,2017) was performed (Supplementary Table S8). The results showed that environmental factors including temperature, salinity, depth, and nitrate, phosphate,and silicate content exhibited a signif icant inf luence on bacterial and archaeal communities (P<0.05). In contrast, only temperature and nitrate exhibited signif icant eff ects on the fungal community (P<0.05).
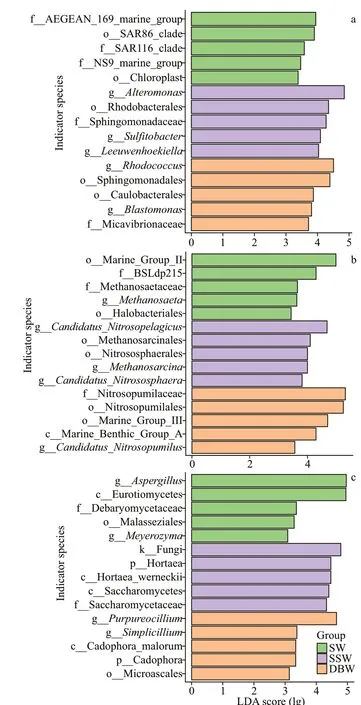
Fig.5 The top f ive indicator species in diff erent water masses on NER by LEfSe analysis
The bacterial communities in SW were signif icantly inf luenced by temperature (Fig.4a;P=0.001)(Supplementary Table S8;P<0.05). At the global scale, positive correlations between bacterial richness and sea-surface temperature have been observed(Pommier et al., 2007; Fuhrman et al., 2015).Sunagawa et al. (2015) demonstrated that vertical stratif ication of epipelagic and mesopelagic bacterial communities was mostly driven by temperature using metagenomic data from 68 diff erent sites across diff erent oceans. However, the SSW bacterial community was strongly inf luenced by salinity(Fig.4a;P=0.001) (Supplementary Table S8;P<0.05).The salinity in SSW changed in very broad range, so that only microbes capable of effi ciently adapting to the shifting salinity could survive (Kuchi and Khandeparker, 2020). The bacterial communities in MW, IW, and DBW exhibited stronger relationships with combined parameters, including depth and nitrate, phosphate, and silicate content (Fig.4a;P=0.001) (Supplementary Table S8;P<0.05). Since temperature and salinity are relatively constant in deeper water masses, including MW, IW, and DBW,the bacterial community is more susceptible to the changing parameters including pressure (depth) and nutrients (Gao et al., 2021). Consequently, the environmental factors (temperature, salinity, depth,and nitrate, phosphate, and silicate content) exerted signif icant impacts on bacterial communities in various water masses.
The environmental parameters aff ecting the archaeal community structure in each water mass were identical to those of bacteria (Fig.4a-b;P=0.001). We speculated that prokaryotic species shared similar adaptation mechanisms to change in the environment in water masses on NER (Gao et al., 2021).
The RDA results clearly demonstrated that single or multiple combined environmental factors altered the prokaryotic community structures in diff erent water masses on NER. Here, temperature was a key environmental variable aff ecting the prokaryotic communities in SW, salinity was a key environmental variable aff ecting the prokaryotic communities in SSW, and combined environmental parameters(including depth and nitrate, phosphate, and silicate content) played a pivotal role in aff ecting the prokaryotic communities in deeper water masses,including MW, IW, and DBW.
3.5 Indicator species of the microbial communities in diff erent water masses on NER
The standardized heatmap presents the top 30 most abundant microbial genera in each water mass on NER (Supplementary Fig.S1). Remarkably, each water mass was distinguished by a phylogenetically close cluster of genera, the abundance of which was higher than that of the rest water masses. In contrast,the genera of high abundance derived from diff erent water masses were phylogenetically distant from each other. Therefore, these results suggested the presence of signature microbial taxa in each water mass on NER. Furthermore, LEfSe and cladogram analyses were performed to determine the indicator species of bacteria, archaea, and fungi in SW, SSW, and DBW,respectively (Fig.5, Supplementary Fig.S2).
With respect to bacteria, the AEGEAN 169 marine group, SAR116 clade, and NS9 marine group were the main indicator species in SW (Fig.5a).Alteromonas,Leeuwenhoekiella,Sulf itobacter, and Sphingomonadaceae were the main indicator species in SSW.Rhodococcus, Micavibvionaceae, andBlastomonaswere the main indicator species in DBW.The indicator species of each water mass adapt well to the environmental conditions of the water mass in which they reside. For example, the AEGEAN 169 marine group exhibits a potentially interesting adaptation to high solar irradiance (Reintjes et al.,2019), the abundance of which is closely related to phytoplanktons (Yang et al., 2015).Alteromonasexhibits relatively strong tolerance to salinity, so it could adapt to the SSW environment (Qin et al.,2020). In addition, strains ofRhodococcushave been characterized as piezophiles (Picard and Daniel,2013). Therefore, this genus could adapt to hostile deep-sea environments with high hydrostatic pressure(Hackbusch et al., 2020).
Among the archaea, BSLdp215 and Methanosaetaceae were the main indicator species in SW (Fig.5b).CandidatusNitrosopelagicus,CanidatusNitrososphaera, andMethanosarcinawere the main indicator species in SSW.CanidatusNitrosopumilusand Nitrosopumilaceae were the main indicator species in DBW. The archaeal indicator species in each water mass are also well-adapted to the physicochemical conditions of the corresponding water mass. For instance,CandidatusNitrosopelagicuscan grow chemolithoautotrophically by aerobically oxidizing ammonia to nitrite (Brochier-Armanet et al., 2008). Thus, aerobic ammonia oxidation appears to be an important pathway in nitrogen cycling in SSW. In addition, several studies have demonstrated that Nitrosopumilaceae are more frequently recovered from deep water (Wemheuer et al., 2019).
With respect to fungi, Debaryomycetaceae was the main indicator species in SW (Fig.5c).Saccharomycetaceae was the main indicator species in SSW.SimpcicilliumandPurpureocilliumwere the main indicator species in DBW. Again, the fungal indicator species adapt well to the environmental conditions of each water mass, but there are fewer indicator species at the levels of family and genus.
In short, LEfSe and cladogram analyses further suggested that diff erent water masses on NER harbor distinct microbial taxa that represent the biological features of the corresponding water mass to some extent. The indicator species dwelling in each water mass might be used as f ingerprints to discriminate corresponding water masses on NER. Though further studies are needed to reinforce this assertion, the present study presented a potential method to identify oceanic water masses using microbiology.
4 CONCLUSION
In the present study, we sorted 60 pelagic water samples on NER into f ive water masses using featuredT-Scurves. The vertical stratif ication and diversity of microbial communities in each water mass were characterized. Bacteria exhibited the highest richness and diversity across the water masses on NER. RDA results suggested that environmental factors in the water masses might play a pivotal role in the formation and stability of the distinct prokaryotic community structure in each water mass. LEfSe analysis further revealed that each water mass harbored distinct indicator species. These indicator species might be used as potential signatures to identify the corresponding water masses on NER. These results substantially reinforced the biological nature of pelagic water masses from the perspective of microbiology. In conclusion, it is robust and feasible to study the vertical distribution, structure, and diversity of microorganisms in pelagic ecosystems based on water masses.
5 DATA AVAILABILITY STATEMENT
The NovaSeq FASTQ f iles and the identif ier barcode f iles were deposited in the National Center for Biotechnology Information Sequence Read Archive (SRA) under BioProject Accession No.PRJNA655383 (https://www.ncbi.nlm.nih.gov/sra/PRJNA655383).
6 ACKNOWLEDGMENT
This is the Key Laboratory of Marine Chemistry Theory and Technology (Ocean University of China),Ministry of Education (MCTL) (Contribution No.249).
 Journal of Oceanology and Limnology2022年2期
Journal of Oceanology and Limnology2022年2期
- Journal of Oceanology and Limnology的其它文章
- Identif ication of Antarctic minke and killer whales with passive acoustic monitoring in Prydz Bay, Antarctica*
- Eff ects of dissolved oxygen and nutrients from the Kuroshio on hypoxia off the Changjiang River estuary*
- Methane in the Yellow Sea and East China Sea: dynamics,distribution, and production*
- Longitudinal genetic analysis of growth-related traits in red swamp crayf ish Procambarus clarkii (Girard)*
- Early life migration and population discrimination of the small yellow croaker Larimichthys polyactis from the Yellow Sea: inferences from otolith Sr/Ca ratios*
- A new oil spill detection algorithm based on Dempster-Shafer evidence theory*