
补益强心片质量标准的改进研究
2022-04-07 03:05刘洪超于雅萌郭东晓林永强
药学研究 2022年3期
刘洪超,于雅萌,郭东晓,林永强
(山东省食品药品检验研究院,国家药品监督管理局胶类产品质量评价重点实验室,山东省中药标准创新与质量评价工程实验室,山东 济南 250101)
补益强心片为人参、丹参、黄芪、麦冬、香加皮、葶苈子等6味中药制备而成的复方制剂,具益气活血的功效,临床用于慢性充血性心力衰竭疗效显著[1]。现行标准收载内容包括性状、人参(人参皂苷Rg1、Re、Rb1)、黄芪(黄芪甲苷)、香加皮(4-甲氧基水杨醛)的薄层色谱(TLC)鉴别及丹参中丹参酮ⅡA的高效液相色谱(HPLC)法含量测定。本试验优化了黄芪和香加皮TLC鉴别的前处理及展开系统,补充了丹参中丹酚酸B的TLC鉴别,增加了HPLC法测定了方中补益元气主药人参中的3种皂苷成分,为该制剂的质量控制提供更为全面准确的方法。
1 材料
1.1 仪器 美国安捷伦1200型高效液相色谱仪;瑞士梅特勒-托利多XSE205型电子天平;昆山市超声KQ-500DE型数控超声波清洗器(500 W,40 kHz);瑞士卡玛TLC Visualizer 2薄层成像系统。
1.2 试药 乙腈为色谱纯,纯化水由密理博 Synergy UV净水系统制备,其他试剂及试药为分析纯;市售硅胶G薄层预制板(青岛海洋化工厂、德国默克公司)。
1.3 标准物质及样品 黄芪甲苷(批号:110781-200613),丹酚酸B(批号:111562-201313),4-甲氧基水杨醛(批号:110790-201303),人参皂苷Rg1(批号:110703-201530),人参皂苷Rb1(批号:110704-201625),人参皂苷Re(批号:110754-201525),丹参对照药材(批号:120923-201113),以上均购于中国食品药品检验研究院。补益强心片4批次(青岛华仁太医药业有限公司,批号:150601、150701、150801、170102),阴性样品均为按处方同比例除去相应药味后制备。
2 方法及结果
2.1 薄层色谱鉴别
2.1.1 黄芪TLC鉴别[2-3]取供试品10片,去除包衣,研细,加甲醇60 mL,回流提取2 h,滤过,甲醇蒸干,残渣加适量水溶解,用水饱和的正丁醇振摇提取3次(20、10、10 mL),合并正丁醇液,用正丁醇饱和的水洗至水层无色,分离正丁醇层蒸干,残渣用约2 mL甲醇溶解,上样于中性氧化铝柱(100至200目,5 g,内径为10~15 mm)上,用100 mL 40%甲醇洗脱,洗脱液浓缩至干,加甲醇1mL溶解,作为供试品溶液。同时取与供试品量相当的不含黄芪阴性样品,按以上供试品制法制成缺黄芪阴性对照溶液。另取黄芪甲苷对照品以甲醇溶解,制成1 mg·mL-1的对照品溶液。分别取供试品溶液、阴性对照溶液各5 μL,对照品溶液2 μL,点样于硅胶G薄层板上,用三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)10 ℃以下放置后分取的下层展开,取出晾干后喷以10%的硫酸乙醇溶液,105 ℃烘至显色清晰。供试品与对照品在相应位置上,显示颜色一致的斑点;在紫外光(365 nm)下检视,斑点显橘黄色荧光。阴性样品在与黄芪甲苷相应色谱位置无斑点和荧光斑点干扰,表明该方法专属性良好,结果见图1。
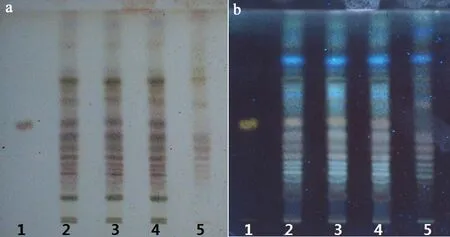
1.黄芪甲苷对照品;2~4.供试品;5.缺黄芪阴性对照 a.日光下;b.紫外光(365 nm)下图1 黄芪薄层鉴别色谱图
2.1.2 香加皮TLC鉴别[2,4]取供试品10片,去除包衣,研细,加甲醇30 mL,回流提取1 h,放冷,滤过蒸干,残渣用甲醇1 mL溶解,作为供试品溶液。同时取与供试品量相当的不含香加皮阴性样品,按以上供试品制法制成缺香加皮阴性对照溶液。另取香加皮对照药材1 g,同供试品制法处理,制成对照药材溶液。再取4-甲氧基水杨醛对照品以甲醇溶解,制成1 mg·mL-1的对照品溶液。取供试品溶液、阴性对照溶液各5 μL,对照药材溶液、对照品溶液各2 μL,点样于硅胶G薄层板上,用石油醚(60~90 ℃)-乙酸乙酯-冰醋酸(20∶3∶0.5)展开,取出晾干后喷以二硝基苯肼试液。供试品在与对照药材相应位置,显相同颜色的多个斑点;在与对照品相应位置,显相同颜色的斑点。阴性样品在香加皮对照药材和4-甲氧基水杨醛对照品相应位置上无斑点干扰,表明该法专属性良好,结果见图2。
2.1.3 丹参TLC鉴别[2,5-6]取供试品10片,去除包衣,研细,加甲醇50 mL,回流提取30 min,放冷,滤过蒸干,残渣加水约40 mL溶解,用石油醚(60~90 ℃)40 mL振摇提取,分取水层,稀盐酸调pH值2~3,加乙醚振摇提取2次,每次30 mL,合并乙醚液,挥去溶剂,加甲醇1 mL溶解,制成供试品溶液。同时取与供试品量相当的不含丹参阴性样品,按以上供试品制法制成缺丹参阴性对照溶液。另取丹参对照药材0.5 g,用20 mL甲醇超声处理20 min,滤过浓缩至1 mL后制成对照药材溶液。再取丹酚酸B对照品以甲醇溶解,制成0.5 mg·mL-1的对照品溶液。分别取供试品溶液、阴性对照溶液、对照药材溶液和对照品溶液各10 μL,点样于硅胶G薄层板上,用甲苯-三氯甲烷-乙酸乙酯-甲醇-甲酸(2∶3∶4∶0.5∶2)展开,取出晾干后喷以5%三氯化铁乙醇溶液。供试品在与对照药材相应位置,显相同颜色的数个斑点;在与对照品相应位置,显相同颜色的斑点。阴性样品在与丹参对照药材和丹酚酸B对照品相应位置上无斑点干扰,表明该法专属性良好,结果见图3。
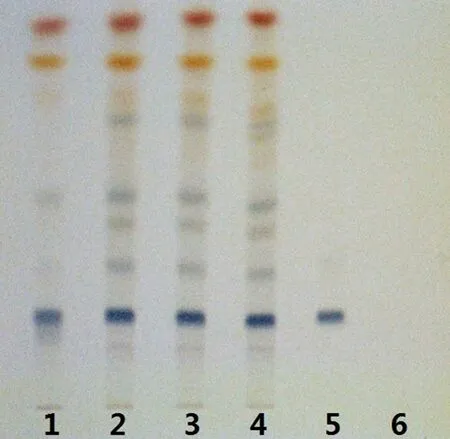
1.丹参对照药材;2~4.供试品;5.丹酚酸B对照品;6.缺丹参阴性对照图3 丹参薄层鉴别色谱图
2.2 含量测定[7-10]
2.2.1 色谱条件 Thermo BDS Hypersil C18色谱柱(4.6 mm×250 mm,5 μm);以乙腈为流动相A,以水为流动相B,梯度洗脱(0~35 min,19%A;35~55 min,19%→29%A;55~70 min,29%A;70~100 min,29%→40%A);流速1.0 mL·min-1,柱温为30 ℃,203 nm处检测;进样体积10 μL。
2.2.2 对照品配制 取人参皂苷Rg1、人参皂苷Re、人参皂苷Rb13种对照品适量,以甲醇溶解,制成含人参皂苷Rg10.187 4 mg·mL-1、人参皂苷Re 0.189 4 mg·mL-1、人参皂苷Rb10.197 2 mg·mL-1的混合对照品溶液;另取适量以上对照品,以甲醇溶解,制成含人参皂苷Rg11.006 9 mg·mL-1、人参皂苷Re 0.935 9 mg·mL-1、人参皂苷Rb10.971 8 mg·mL-1的混合对照品储备液,用于线性关系的测定。
2.2.3 供试品制备 取供试品20片,去除包衣,研细,取约2 g于滤纸筒内,精密称定,置索氏提取器中,加适量三氯甲烷回流至提取液无颜色,弃去提取液,残渣挥干溶剂,连同滤纸筒移入烧瓶中,精密加70%乙醇50 mL,称定重量,回流提取1 h,放冷,用70%乙醇补足失重,摇匀,滤过,精密移取25 mL续滤液至蒸发皿中,蒸去溶剂,用30 mL水分次溶解残渣,转移至分液漏斗中,水饱和正丁醇振摇提取4次(30 mL×2、20 mL×2),正丁醇提取液合并,以氨试液30 mL洗涤2次,弃去氨试液,回收正丁醇至干,残渣以甲醇溶解转移至5 mL量瓶中,定容,摇匀,滤过即得。
2.2.4 阴性对照制备 取相当于供试品量约2 g的不含人参阴性样品,按以上供试品制法制成缺人参阴性对照溶液。
2.2.5 系统适用性 取以上对照品和供试品溶液各10 μL,按“2.2.1”项下色谱条件,进样并记录相关色谱信息。理论板数按人参皂苷Rg1、人参皂苷Re和人参皂苷Rb1峰计均不低于6 000;目标峰基线分离良好,与相邻色谱峰分离度均大于1.5。
2.2.6 线性关系 精密吸取“2.2.2”项下的对照品溶液1、2、5 mL分别置10 mL量瓶中,加甲醇稀释至刻度,摇匀;再取对照品储备液5 mL置10 mL量瓶中,加甲醇稀释至刻度,摇匀。取以上4份稀释溶液及“2.2.2”项下的对照品溶液、对照品储备液各进样10 μL,测定并记录3个目标峰的峰面积。每个对照品均以进样量对应横坐标,峰面积对应纵坐标,进行线性回归分析,得到如下结果:人参皂苷Rg1回归方程Y=341.52X+5.214 4,相关系数r=1.000 0;人参皂苷Re回归方程Y=323.66X-13.611,相关系数r=0.999 9;人参皂苷Rb1回归方程Y=239.7X+14.789,相关系数r=0.999 6。试验结果表明,3种人参皂苷对照品进样量分别在0.187 4~10.06 9 μg(人参皂苷Rg1)、0.1894~9.359 μg(人参皂苷Re)、0.197 2~9.718 μg(人参皂苷Rb1)范围内与所测得的峰面积积分值线性关系良好。
2.2.7 精密度试验 取“2.2.2”项下混合对照品溶液10 μL,注入液相色谱仪,连续进样6次,测其峰面积积分值。结果3种目标成分的峰面积RSD分别为0.45%(人参皂苷Rg1)、0.31%(人参皂苷Re)、0.38%(人参皂苷Rb1),结果显示仪器精密度良好。
2.2.8 稳定性试验 取“2.2.3”项下的供试品,在制备后的0、2、4、8、12、24 h时间点各进样10 μL,对3种目标成分的峰面积进行测定。结果3种目标成分的峰面积RSD分别为1.04%(人参皂苷Rg1)、0.44%(人参皂苷Re)、0.64%(人参皂苷Rb1),表明供试品中所测成分在24 h内有良好的稳定性。
2.2.9 重复性考察 取批号为170102的样品,照“2.2.3”项下所述方法处理,平行制备6份,所得供试品溶液依“2.2.1”项下色谱条件测定,计算含量。结果人参皂苷Rg1的平均含量为0.571 6 mg·g-1(RSD为 0.45%,n=6)、人参皂苷Re的平均含量为0.777 4 mg·g-1(RSD为0.86%,n=6)、人参皂苷Rb1的平均含量为1.433 7 mg·g-1(RSD为0.46%,n=6),表明拟定的检测方法重复性良好。
2.2.10 准确度试验 取已在重复性试验中测定含量的170102批次样品1.0 g,按“2.2.3”项下供试品制备方法操作至“连同滤纸筒移入具塞锥形瓶中”,精密加入对照品溶液(浓度为:人参皂苷Rg10.052 42 mg·mL-1、人参皂苷Re 0.067 45 mg·mL-1、人参皂苷Rb10.127 68 mg·mL-1,70%乙醇配制)10 mL和70%乙醇40 mL,继按“2.2.3”项下所述供试品溶液制备方法处理至制备结束,平行制备6份。计算每份样品中3种目标成分回收率,结果见表1。
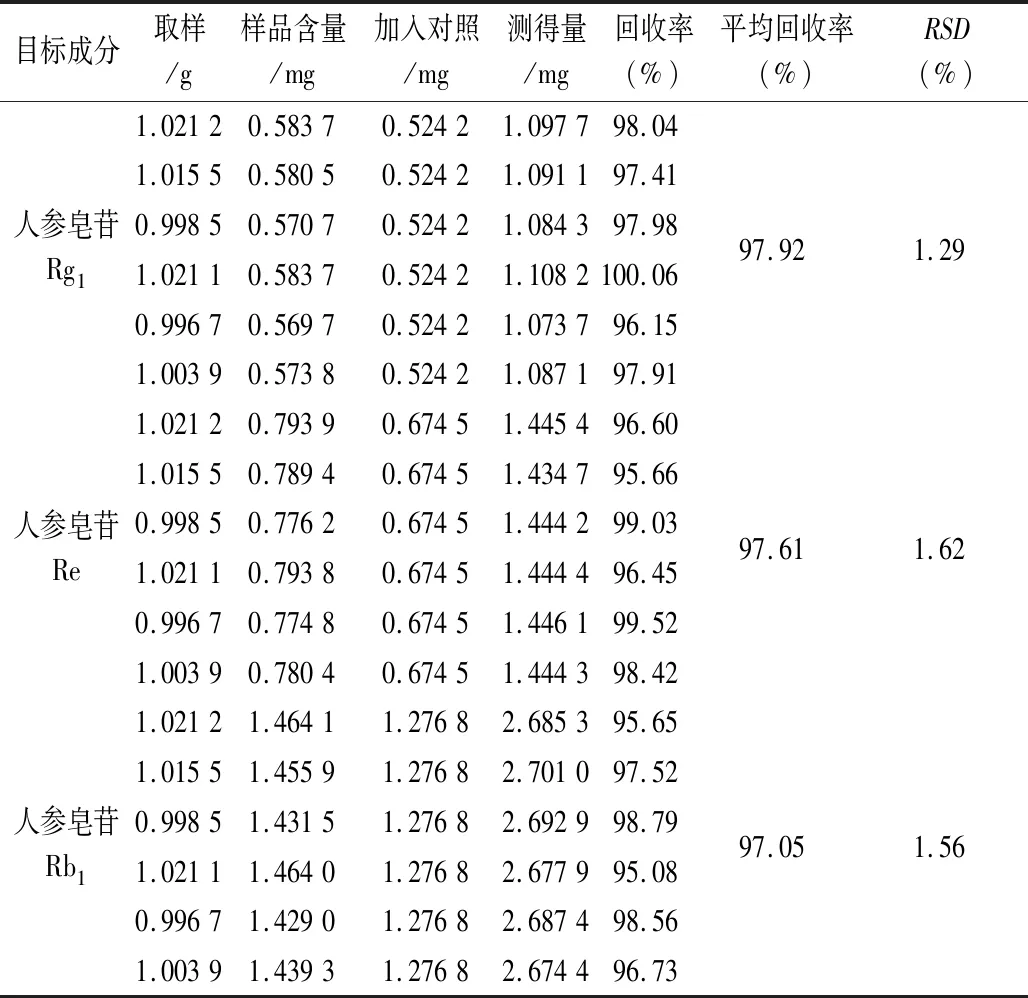
表1 准确度试验结果(n=6)
2.2.11 专属性试验 取“2.2.2”项下对照品溶液、“2.2.3”项下供试品溶液及“2.2.4”项下缺人参阴性对照溶液,照“2.2.1”项下色谱条件,分别进样10 μL,记录相关色谱信息。供试品色谱中呈现与对照品色谱相应的3个目标峰,阴性样品色谱中无相应色谱峰,证明处方中其他药味对测定人参中的目标成分无干扰(见图4)。
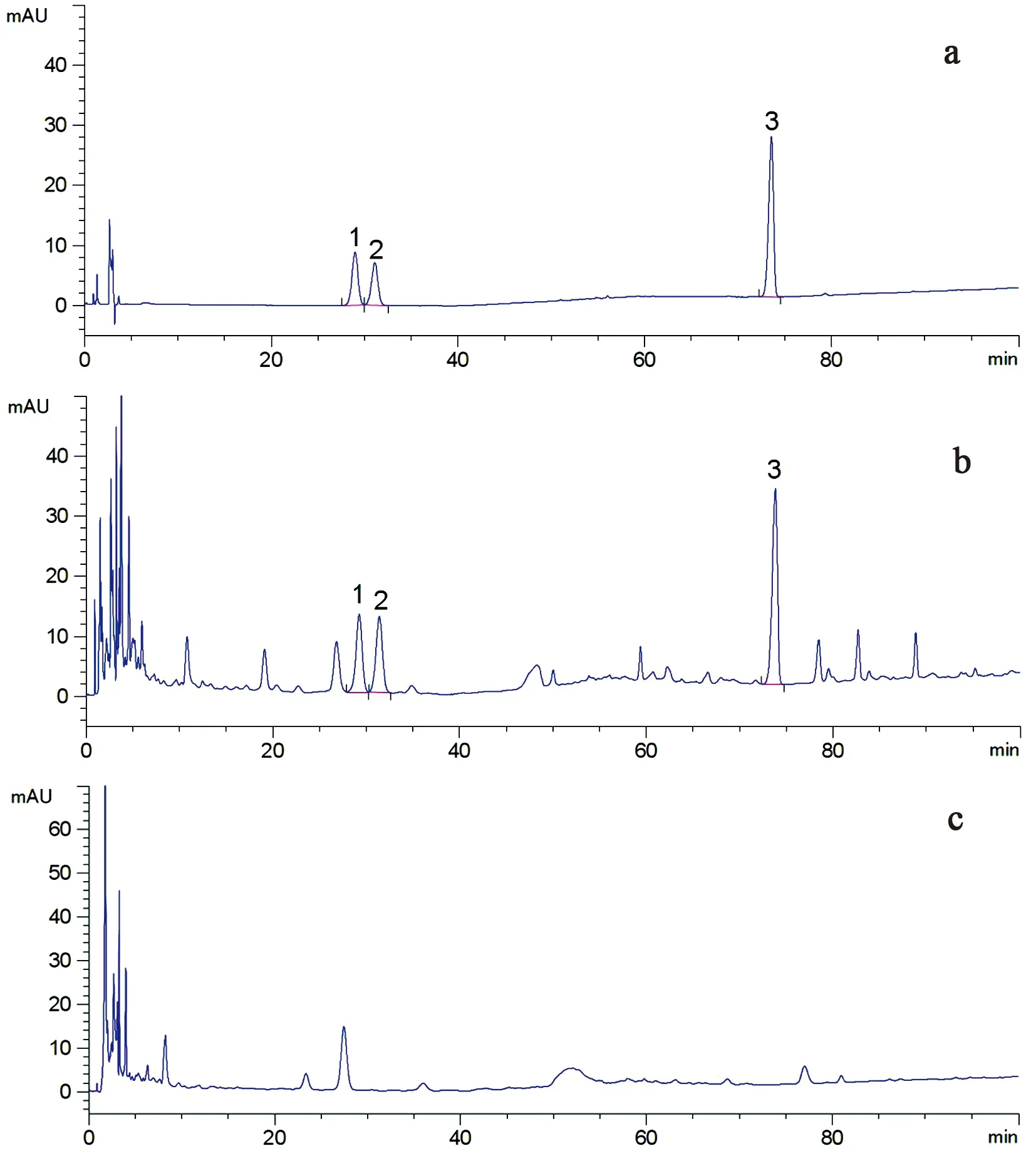
a.混合对照品溶液;b.供试品溶液;c.阴性对照溶液 1.人参皂苷Rg1;2.人参皂苷Re;3.人参皂苷Rb1图4 人参含量测定HPLC色谱图
2.2.12 样品测定 取4批样品各2份,按照“2.2.3”项下的方法进行测定,以外标法计算人参皂苷Rg1、Re、Rb1的含量总和,结果见表2。
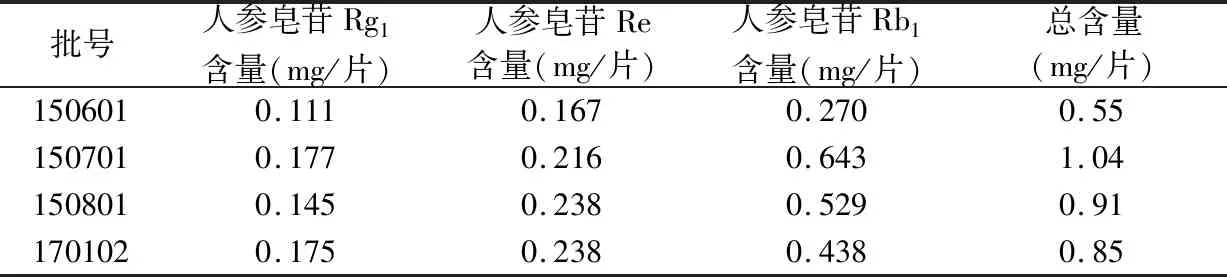
表2 供试品含量测定结果(n=2)
3 讨论
3.1 薄层鉴别 黄芪甲苷鉴别原标准中提取较为烦琐,上大孔树脂柱后需反复冲洗,后续还需进行索氏提取,改为萃取后蒸干过中性氧化铝柱进行除杂纯化,更为便捷省时,且分离较好无干扰。优化了香加皮薄层鉴别项的展开剂及显色剂,并增加了香加皮药材作为对照,可在TLC图谱中体现更多对应的色谱信息。增加了丹参对照药材和丹酚酸B的薄层鉴别,可对原标准中丹参的质量控制(丹参酮ⅡA的含量测定)予以补充。以上各鉴别均进行了阴性实验及耐用性实验,结果均满足TLC方法的相关要求。
3.2 补益强心片原质量标准中包含丹参中丹参酮ⅡA的HPLC法含量测定,属于丹参中脂溶性有效成分,辅以新增的丹参对照药材及水溶性成分丹酚酸B的TLC鉴别,可以有效控制药品中丹参的质量。
3.3 提取方法的优化 新增人参中3种人参皂苷的含量测定,并对提取条件进行了考察和优化。以70%乙醇作为提取溶剂同等条件下效率明显高于甲醇,加热回流效率略高于超声;3种目标成分均为中性人参皂苷,水饱和正丁醇4次萃取基本实现全部转移;除杂方法考察了浓氨试液和氨试液,二者较NaOH溶液可实现洗涤后直接蒸干,减少水洗去除目标部位残留不挥发碱的步骤,其2次萃取后均能基本去除干扰,考虑到操作时的强刺激性,确定使用氨试液作为洗涤溶剂。另对取样量、提取时间、萃取次数等条件进行了考察,最终确定了“2.2.3”项下的样品提取方法。
3.4 色谱条件的优化 3种待测人参皂苷最大吸收波长均在(203±2)nm,该区域靠近紫外末端吸收区,干扰较大,故流动相的优选较为必要。在尝试数种更为快速的色谱条件未果后,最终确定参照药典中人参药材项下色谱条件进行试验,运行时间稍长但可将目标成分有效分离,满足试验需要。另对多根不同品牌及填料色谱柱(Thermo BDS Hypersil;Waters Symmertry;Kromasil 100-5,规格均为C18,4.6 mm×250 mm,5 μm)进行了考察,耐用性均满足试验需要。
本研究采用更为便捷、专属且重现性好的方法可以对补益强心片中相关指标进行有效检测,可为其质量标准的提高提供依据。
猜你喜欢
今日农业(2022年4期)2022-11-16
中医药导报(2022年6期)2022-11-07
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
Digital Chinese Medicine(2021年2期)2022-01-19
河南农业·综合版(2021年7期)2021-08-23
河南农业(2021年7期)2021-07-30
Digital Chinese Medicine(2021年3期)2021-07-23
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
